Biology:2 base encoding
2 Base Encoding, also called SOLiD (sequencing by oligonucleotide ligation and detection), is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing (introduced in 2005), the Solexa system (introduced in 2006) and the SOLiD system (introduced in 2007). These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.
2-base encoding is based on ligation sequencing rather than sequencing by synthesis.[1] However, instead of using fluorescent labeled 9-mer probes that distinguish only 6 bases, 2-base encoding takes advantage of fluorescent labeled 8-mer probes that distinguish the two 3 prime most bases but can be cycled similar to the Macevicz method, thus greater than 6bp reads can be obtained (25-50bp published,[2] 50bp in NCBI in Feb 2008). The 2 base encoding enables reading each base twice without performing twice the work.[3][4][5][6]
General features
The general steps common to many of these next-generation sequencing techniques include:
- Random fragmentation of genomic DNA
- Immobilization of single DNA fragments on a solid support like a bead or a planar solid surface
- Amplification of DNA fragments on the solid surface using PCR and making polymerase colonies[7]
- Sequencing and subsequent in situ interrogation after each cycle using fluorescence scanning or chemiluminescence.[8]
In 1988, Whiteley et al. demonstrated the use of fluorescently labeled oligonucleotide ligation for the detection of DNA variants.[9] In 1995 Macevicz[10] demonstrated repeated ligation of oligonucleotides to detect contiguous DNA variants. In 2003, Dressman et al.[11] demonstrated the use of emulsion PCR to generate millions of clonally amplified beads which one could perform these repeated ligation assays on. In 2005, Shendure et al. performed a sequencing procedure which combined Whiteley and Dressman techniques performing ligation of fluorescent labeled "8 base degenerate" 9-mer probes which distinguished a different base according to the probes label and non degenerate base. This process was repeated (without regenerating an extendable end as in Macevicz) using identical primers but with probes with labels which identified different non-degenerate base to sequence 6bp reads in 5->3 direction and 7bp reads in the 3->5 direction.
How it works
The SOLiD Sequencing System uses probes with dual base encoding.
The underlying chemistry is summarized in the following steps:[12]
- Step 1, Preparing a Library: This step begins with shearing the genomic DNA into small fragments. Then, two different adapters are added (for example A1 and A2). The resulting library contains template DNA fragments, which are tagged with one adapter at each end (A1-template-A2).
- Step 2, Emulsion PCR: In this step, the emulsion (droplets of water suspended in oil) PCR reaction is performed using DNA fragments from library, two primers (P1 and P2) that complement to the previously used adapters (P1 with A1 and P2 with A2), other PCR reaction components and 1μm beads coupled with one of the primers (e.g. P1) make dilution from DNA library to maximize the droplet that contain one DNA fragment and one bead into a single emulsion droplet.
In each droplet, DNA template anneals to the P1-coupled bead from its A1 side. Then DNA polymerase will extend from P1 to make the complementary sequence, which eventually results in a bead enriched with PCR products from a single template. After PCR reaction, templates are denatured and disassociate from the beads. Dressman et al. first describe this technique in 2003.
- Step 3, Bead Enrichment: In practice, only 30% of beads have target DNA. To increase the number of beads that have target DNA, large polystyrene beads coated with A2 are added to the solution. Thus, any bead containing the extended products will bind polystyrene bead through its P2 end. The resulting complex will be separated from untargeted beads, and melt off to dissociate the targeted beads from polystyrene. This step can increase the throughput of this system from 30% before enrichment to 80% after enrichment.
After enrichment, the 3’-end of products (P2 end) will be modified which makes them capable of covalent bonding in the next step. Therefore, the products of this step are DNA-coupled beads with 3’-modification of each DNA strand.
- Step 4, Bead Deposition: In this step, products of the last step are deposited onto a glass slide. Beads attach to the glass surface randomly through covalent bonds of the 3’-modified beads and the glass.
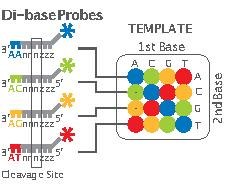
- Step 5, Sequencing Reaction: As mentioned earlier, unlike other next-generation methods which perform sequencing through synthesis, 2-base encoding is based on sequencing by ligation. The ligation is performed using specific 8-mer probes:
These probes are eight bases in length with a free hydroxyl group at the 3’ end, a fluorescent dye at the 5’ end and a cleavage site between the fifth and sixth nucleotide. The first two bases (starting at the 3' end) are complementary to the nucleotides being sequenced. Bases 3 through 5 are degenerate and able to pair with any nucleotides on the template sequence. Bases 6-8 are also degenerate but are cleaved off, along with the fluorescent dye, as the reaction continues. Cleavage of the fluorescent dye and bases 6-8 leaves a free 5' phosphate group ready for further ligation. In this manner positions n+1 and n+2 are correctly base-paired followed by n+6 and n+7 being correctly paired, etc. The composition of bases n+3, n+4 and n+5 remains undetermined until further rounds of the sequencing reaction.
The sequencing step is basically composed of five rounds and each round consists of about 5-7 cycles (Figure 2). Each round begins with the addition of a P1-complementary universal primer. This primer has, for example, n nucleotides and its 5’-end matches exactly with the 3’-end of the P1. In each cycle, 8-mer probes are added and ligated according to their first and second bases. Then, the remaining unbound probes are washed out, the fluorescent signal from the bound probe is measured, and the bound probe is cleaved between its fifth and sixth nucleotide. Finally the primer and probes are all reset for the next round.
In the next round a new universal primer anneals the position n-1 (its 5’-end matches to the base exactly before the 3’-end of the P1) and the subsequent cycles are repeated similar to the first round. The remaining three rounds will be performed with new universal primers annealing positions n-2, n-3 and n-4 relative to the 3'-end of P1.
A complete reaction of five rounds allows the sequencing of about 25 base pairs of the template from P1.
- Step 6, Decoding Data: For decoding the data, which are represented as colors, we must first know two important factors. First, we must know that each color indicates two bases. Second, we need to know one of the bases in the sequence: this base is incorporated in the sequence in the last (fifth) round of step5. This known base is the last nucleotide of the 3’-end of the known P1. Therefore, since each color represents two nucleotides in which the second base of each dinucleotide unit constitutes the first base of the following dinucleotide, knowing just one base in the sequence will lead us to interpret the whole sequence(Figure 2).[13]
2 Base Encoding considerations
In practice direct translation of color reads into base reads is not advised as the moment one encounters an error in the color calls it will result in a frameshift of the base calls. To best leverage the "error correction" properties of two base encoding it is best to convert your base reference sequence into color-space. There is one unambiguous conversion of a base reference sequence into color-space and while the reverse is also true the conversion can be wildly inaccurate if there are any sequencing errors.[14]
Mapping color-space reads to a color-space reference can properly utilize the two-base encoding rules where only adjacent color differences can represent a true base polymorphism. Direct decoding or translation of the color reads into bases cannot do this efficiently without other knowledge.
More specifically, this method is not an error correction tool but an error transformation tool. Color-space transforms your most common error mode (single measurement errors) into a different frequency than your most common form of DNA variation (SNPs or single base changes). These single base changes affect adjacent colors in color space. There are logical rules which help correct adjacent errors into 'valid' and 'invalid' adjacent errors.
The likelihood of getting two adjacent errors in a 50-bp read can be estimated. There are 49 ways of making adjacent changes to a 50 letter string (50-bp read). There are 1225 ways of making non-adjacent changes to a 50 letter string (50 choose 2). Simplistically, if one assumes errors are completely random (they are usually higher frequency at the end of reads) only 49 out of 1225 errors will be candidates for SNPs. In addition, only one third of the adjacent errors can be valid errors according to the known labeling of the probes thus delivering only 16 out of 1225 errors which can be candidates for SNPs. This is particularly useful for low coverage SNP detection as it reduces false positives at low coverage, Smith et al.[15]
Advantages
Each base in this sequencing method is read twice. This changes the color of two adjacent color space calls, therefore in order to miscall a SNP, two adjacent colors must be miscalled. Because of this the SNP miscall rate is on the order of e^2, where e is the device error rate.
Disadvantages
When base calling single color miscalls cause errors on the remaining portion of the read. In SNP calling this can be corrected, which results in a lower SNP calling error rate. However for simplistic de novo assembly you are left with the raw device error rate which will be significantly higher than the 0.06% reported for SNP calling. Quality filtering of the reads can deliver higher raw accuracy reads which when aligned to form color contigs can deliver reference sequences where 2 base encoding can be better leveraged. Hybrid assemblies with other technologies can also better utilize the 2 base encoding.
See also
- DNA sequencing
- Sequencing by ligation
- Single Molecule Real Time Sequencing
References
- ↑ Jay Shendure et al. (2005) Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 309(5741), 1728 - 1732
- ↑ Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, Zhang Z, Ranade SS, Dimalanta ET, Hyland FC, Sokolsky TD, Zhang L, Sheridan A, Fu H, Hendrickson CL, Li B, Kotler L, Stuart JR, Malek JA, Manning JM, Antipova AA, Perez DS, Moore MP, Hayashibara KC, Lyons MR, Beaudoin RE, Coleman BE, Laptewicz MW, Sannicandro AE, Rhodes MD, Gottimukkala RK, Yang S, Bafna V, Bashir A, MacBride A, Alkan C, Kidd JM, Eichler EE, Reese MG, De La Vega FM, Blanchard AP. Genome Res. 2009 Sep;19(9):1527-41. Epub 2009 Jun 22.
- ↑ Patent: Reagents,Methods and Libraries for Bead-Based Sequencing
- ↑ Article: A high-resolution, nucleosome position map of C. elegans reveals a lack of universal...
- ↑ Article: Stem cell transcriptome profiling via massive-scale mRNA sequencing
- ↑ Rapid whole-genome mutational profiling using next-generation sequencing technologies, Genome Research, 2008 18:1638-1642
- ↑ Chetverin, NAR, 1993, Vol.21, No. 10 2349-2353
- ↑ MATTHEW E. HUDSON (2008) Sequencing breakthroughs for genomic ecology and evolutionary biology. Molecular Ecology Resources 8 (1), 3–17
- ↑ Whiteley US patent number 4,883,750
- ↑ Macevicz US patent number 5,750,341
- ↑ Transforming single DNA molecules into fluorescent magnetic particles fr detection and enumeration of genetic variations,PNAS July 22, 2004 Vol. 100 no. 15, 8817-8822
- ↑ Applied Biosystems
- ↑ Tech Summary: ABI's SOLiD (Seq. by Oligo Ligation/Detection) - SEQanswers
- ↑ [1] Colorspace to FastQ example
- ↑ Smith et al., Genome Research 2008 18:1638-1642
 |  |

