Biology:ATAC-seq
ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is a laboratory technique used in molecular biology to assess genome-wide chromatin accessibility.[1] The technique was introduced in 2013 by the labs of Will Greenleaf and Howard Chang at Stanford University as an alternative to MNase-seq, FAIRE-Seq and DNase-Seq[1] with faster turnaround time, simpler protocol, and lower DNA input requirements.[2][3][4]
Procedure
ATAC-seq identifies accessible DNA regions by probing open chromatin with hyperactive mutant Tn5 Transposase that inserts sequencing adapters into open regions of the genome.[2][5] While naturally occurring transposases have a low level of activity, ATAC-seq employs the mutated hyperactive transposase.[6] In a process called "tagmentation", Tn5 transposase cleaves and tags double-stranded DNA with sequencing adaptors in a single enzymatic step.[7][8] The tagged DNA fragments are then purified, PCR-amplified, and sequenced using next-generation sequencing.[8] Sequencing reads can then be used to infer regions of increased accessibility as well as to map regions of transcription factor binding sites and nucleosome positions.[2] The number of reads for a region correlate with how open that chromatin is, at single nucleotide resolution.[2]
ATAC-seq requires no sonication or phenol-chloroform extraction like FAIRE-seq;[9] no antibodies like ChIP-seq;[10] and no sensitive enzymatic digestion like MNase-seq or DNase-seq.[11] ATAC-seq preparation can be completed in under three hours.[12]
Applications
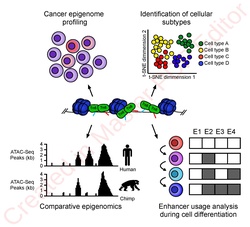
ATAC-Seq analysis is used to investigate a number of chromatin-accessibility signatures. The most common use is nucleosome mapping experiments,[3] but it can be applied to mapping transcription factor binding sites,[13] adapted to map DNA methylation sites,[14] or combined with sequencing techniques.[15]
The utility of high-resolution enhancer mapping ranges from studying the evolutionary divergence of enhancer usage (e.g. between chimps and humans) during development[16] and uncovering a lineage-specific enhancer map used during blood cell differentiation.[17]
ATAC-Seq has also been applied to defining the genome-wide chromatin accessibility landscape in human cancers,[18] and revealing an overall decrease in chromatin accessibility in macular degeneration.[19] Computational footprinting methods can be performed on ATAC-seq to find cell specific binding sites and transcription factors with cell specific activity.[13]
ATAC-seq has found increasing applications in clinical research and disease studies. EPIC-ATAC has been developed as a deconvolution framework to quantify cell-type heterogeneity in bulk tumor ATAC-seq data, enabling analysis of regulatory processes underlying tumor development and correlation with clinical variables in cancer research.[20][21] In immunology, ATAC-seq has been used to characterize dynamic epigenetic changes in T cell exhaustion, revealing that exhausted T cells possess unique chromatin accessibility patterns compared to naive, effector, and memory T cells, with implications for cancer immunotherapy.[22] The Cancer Genome Atlas has generated genome-wide chromatin accessibility profiles of 410 tumor samples spanning 23 cancer types, identifying 562,709 transposase-accessible DNA elements and revealing genetic risk loci of cancer predisposition as active DNA regulatory elements.[23] Integrative analysis combining ATAC-seq with RNA-seq has been used to identify novel oncogenes and elucidate regulatory mechanisms in hepatocellular carcinoma.[24]
Single-cell ATAC-seq
Modifications to the ATAC-seq protocol have been made to accommodate single-cell analysis. Microfluidics can be used to separate single nuclei and perform ATAC-seq reactions individually.[12] With this approach, single cells are captured by either a microfluidic device or a liquid deposition system before tagmentation.[12][25] An alternative technique that does not require single cell isolation is combinatorial cellular indexing.[26] This technique uses barcoding to measure chromatin accessibility in thousands of individual cells; it can generate epigenomic profiles from 10,000-100,000 cells per experiment.[27] But combinatorial cellular indexing requires additional, custom-engineered equipment or a large quantity of custom, modified Tn5.[28] Recently, a pooled barcode method called sci-CAR was developed, allowing joint profiling of chromatin accessibility and gene expression of single cells.[29]
Computational analysis of scATAC-seq is based on construction of a count matrix with number of reads per open chromatin regions. Open chromatin regions can be defined, for example, by standard peak calling of pseudo bulk ATAC-seq data. Further steps include data reduction with PCA and clustering of cells.[25] scATAC-seq matrices can be extremely large (hundreds of thousands of regions) and is extremely sparse, i.e. less than 3% of entries are non-zero.[30] Therefore, imputation of count matrix is another crucial step performed by using various methods such as non-negative matrix factorization. As with bulk ATAC-seq, scATAC-seq allows finding regulators like transcription factors controlling gene expression of cells. This can be achieved by looking at the number of reads around TF motifs[31] or footprinting analysis.[30]
Spatial ATAC-seq
Spatial ATAC-seq combines chromatin accessibility profiling with spatial information, enabling researchers to map epigenetic landscapes while preserving tissue architecture. This method combines in situ Tn5 transposition chemistry with microfluidic deterministic barcoding to perform spatially resolved chromatin accessibility analysis on tissue sections at the cellular level and genome scale.[32][33] The technique has been applied to co-profiling of the epigenome and transcriptome, facilitating investigation of the correlation between accessible peaks and expressed genes pixel by pixel in the tissue context.[32]
Recent developments include SPACE-seq (SPatial assay for Accessible chromatin, Cell lineages, and gene Expression with sequencing), which enables simultaneous analysis of gene expression, chromatin accessibility, and mitochondrial DNA mutations using commercially available spatial transcriptomics platforms.[34] Laser capture microdissection coupled to ATAC-seq (LCM-ATAC-seq) has also been developed for targeted chromatin accessibility analysis of discrete contiguous or scattered cell populations in tissues, enabling analysis at mini-bulk resolution with the possibility to integrate cellular or morphological stainings.[35]
A commercially implemented form of deterministic barcoding for spatial ATAC-seq is provided by AtlasXomics, which commercializes the DBiT-seq microfluidic barcoding technology originally developed in the Fan laboratory.[36] The platform generates ~10 μm spatial pixels through orthogonal microfluidic barcoding flows and is compatible with both fresh-frozen and FFPE (formalin-fixed paraffin-embedded) tissue sections. Peer-reviewed applications of AtlasXomics-based spatial ATAC-seq include mapping the chromatin accessibility landscape of the human dorsal root ganglion using combined spatial ATAC-seq and CUT&Tag.[37] Additional studies using this platform have been reported in preprint form, extending spatial epigenomic analysis to cancer models, developmental tissues, and immune microenvironments.[38][39]
Multimodal ATAC-seq
Recent advances have enabled simultaneous profiling of chromatin accessibility alongside other molecular modalities in the same cells or tissue sections. Spatial ATAC–RNA-seq and spatial CUT&Tag–RNA-seq allow co-profiling of genome-wide chromatin accessibility or histone modifications in conjunction with whole transcriptome on the same tissue section at near-single-cell resolution.[32] ISSAAC-seq (In Situ Sequencing of chromatin Accessibility And Cellular transcriptomes) represents a multimodal update to ATAC-seq, providing a powerful method for investigating gene expression and chromatin accessibility within the same cell at high sensitivity and lower cost than commercially available kits.[40] These multimodal approaches have led to the development of computational tools like SCRIPro, which combines transcription factor-target importance from epigenomic data with transcription factor-target expression from transcriptomic data to construct gene regulatory networks from single-cell and spatial multiomics data.[41]
Computational Tools and Analysis
ATAC-seq data analysis presents unique methodological challenges due to data sparsity and the need for specialized bioinformatics tools. The major steps include pre-analysis (quality check and alignment), core analysis (peak calling), and advanced analysis (peak differential analysis and annotation, motif enrichment, footprinting, and nucleosome position analysis).[42][43] MACS2 (Model-based Analysis of ChIP-seq 2) remains the most widely used peak caller for ATAC-seq data analysis, serving as the default peak caller in the ENCODE ATAC-seq pipeline. Originally developed for ChIP-seq, MACS2 has been adapted for ATAC-seq analysis and performs well for identifying regions of enriched transposase accessibility, though it requires parameter optimization for ATAC-seq-specific characteristics.[44][45] Standardized analysis workflows have been developed, including the nf-core/atacseq pipeline, which provides a comprehensive, reproducible framework for ATAC-seq data processing from raw reads to final peak calls and quality control metrics. This Nextflow-based pipeline incorporates best practices for adapter trimming, alignment, duplicate removal, peak calling, and downstream analysis, facilitating standardized processing across different research groups and computational environments.[46][47]
See also
- ScGET-seq
- Single-cell analysis
- William Greenleaf (American scientist)
- Howard Y. Chang
References
- ↑ 1.0 1.1 "Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position". Nature Methods 10 (12): 1213–8. December 2013. doi:10.1038/nmeth.2688. PMID 24097267. Bibcode: 2013NatCB..10.1213B.
- ↑ 2.0 2.1 2.2 2.3 "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology 109: 21.29.1–21.29.9. January 2015. doi:10.1002/0471142727.mb2129s109. PMID 25559105.
- ↑ 3.0 3.1 "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research 25 (11): 1757–70. November 2015. doi:10.1101/gr.192294.115. PMID 26314830. Bibcode: 2015GenRe..25.1757S.
- ↑ "DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells". Cold Spring Harbor Protocols 2010 (2). February 2010. doi:10.1101/pdb.prot5384. PMID 20150147.
- ↑ Bajic, Marko; Maher, Kelsey A.; Deal, Roger B. (2018). "Identification of Open Chromatin Regions in Plant Genomes Using ATAC-Seq". Plant Chromatin Dynamics. Methods in Molecular Biology. 1675. pp. 183–201. doi:10.1007/978-1-4939-7318-7_12. ISBN 978-1-4939-7317-0.
- ↑ "Transposon Tn5". Annual Review of Genetics 42 (1): 269–86. 2008. doi:10.1146/annurev.genet.42.110807.091656. PMID 18680433.
- ↑ Adey, Andrew (December 2010). "Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition". Genome Biology 11 (12). doi:10.1186/gb-2010-11-12-r119. PMID 21143862. Bibcode: 2010GenBi..11.R119A.
- ↑ 8.0 8.1 "Tn5 transposase and tagmentation procedures for massively scaled sequencing projects". Genome Research 24 (12): 2033–40. December 2014. doi:10.1101/gr.177881.114. PMID 25079858.
- ↑ "Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA". Nature Protocols 7 (2): 256–67. January 2012. doi:10.1038/nprot.2011.444. PMID 22262007.
- ↑ "CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins". Genome Research 25 (10): 1581–9. October 2015. doi:10.1101/gr.193540.115. PMID 26355004.
- ↑ Hoeijmakers, Wieteke Anna Maria; Bártfai, Richárd (2018). "Characterization of the Nucleosome Landscape by Micrococcal Nuclease-Sequencing (MNase-seq)". Chromatin Immunoprecipitation. Methods in Molecular Biology. 1689. pp. 83–101. doi:10.1007/978-1-4939-7380-4_8. ISBN 978-1-4939-7379-8.
- ↑ 12.0 12.1 12.2 "Single-cell chromatin accessibility reveals principles of regulatory variation". Nature 523 (7561): 486–90. July 2015. doi:10.1038/nature14590. PMID 26083756. Bibcode: 2015Natur.523..486B.
- ↑ 13.0 13.1 Li, Zhijian; Schulz, Marcel H.; Look, Thomas; Begemann, Matthias; Zenke, Martin; Costa, Ivan G. (26 February 2019). "Identification of transcription factor binding sites using ATAC-seq". Genome Biology 20 (1): 45. doi:10.1186/s13059-019-1642-2. PMID 30808370.
- ↑ "methyl-ATAC-seq measures DNA methylation at accessible chromatin". Genome Research 29 (6): 969–977. June 2019. doi:10.1101/gr.245399.118. PMID 31160376.
- ↑ Hendrickson, David G.; Soifer, Ilya; Wranik, Bernd J.; Botstein, David; Scott McIsaac, R. (2018), "Simultaneous Profiling of DNA Accessibility and Gene Expression Dynamics with ATAC-Seq and RNA-Seq", Computational Cell Biology, Methods in Molecular Biology, 1819, Springer New York, pp. 317–333, doi:10.1007/978-1-4939-8618-7_15, ISBN 978-1-4939-8617-0, PMID 30421411
- ↑ "Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest". Cell 163 (1): 68–83. September 2015. doi:10.1016/j.cell.2015.08.036. PMID 26365491.
- ↑ "Immunogenetics. Chromatin state dynamics during blood formation". Science 345 (6199): 943–9. August 2014. doi:10.1126/science.1256271. PMID 25103404.
- ↑ "The chromatin accessibility landscape of primary human cancers". Science 362 (6413). October 2018. doi:10.1126/science.aav1898. PMID 30361341. Bibcode: 2018Sci...362.1898C.
- ↑ "ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration". Nature Communications 9 (1). April 2018. doi:10.1038/s41467-018-03856-y. PMID 29636475. Bibcode: 2018NatCo...9.1364W.
- ↑ Gabriel, Aurélie AG; Racle, Julien; Falquet, Maryline; Jandus, Camilla; Gfeller, David (2024). "Robust estimation of cancer and immune cell-type proportions from bulk tumor ATAC-Seq data" (in en). eLife 13. doi:10.7554/eLife.94833.4.
- ↑ Corces, M. Ryan; Granja, Jeffrey M.; Shams, Shadi; Louie, Bryan H.; Seoane, Jose A.; Zhou, Wanding; Silva, Tiago C.; Groeneveld, Clarice et al. (2018-10-26). "The chromatin accessibility landscape of primary human cancers". Science 362 (6413). doi:10.1126/science.aav1898. PMID 30361341. Bibcode: 2018Sci...362.1898C.
- ↑ Chen, Chufeng; Liu, Jiaying; Chen, Yidong; Lin, Anqi; Mou, Weiming; Zhu, Lingxuan; Yang, Tao; Cheng, Quan et al. (January 2023). "Application of ATAC-seq in tumor-specific T cell exhaustion" (in en). Cancer Gene Therapy 30 (1): 1–10. doi:10.1038/s41417-022-00495-w. ISSN 1476-5500. PMID 35794339.
- ↑ Taavitsainen, S.; Engedal, N.; Cao, S.; Handle, F.; Erickson, A.; Prekovic, S.; Wetterskog, D.; Tolonen, T. et al. (2021-09-06). "Single-cell ATAC and RNA sequencing reveal pre-existing and persistent cells associated with prostate cancer relapse" (in en). Nature Communications 12 (1): 5307. doi:10.1038/s41467-021-25624-1. ISSN 2041-1723. PMID 34489465. Bibcode: 2021NatCo..12.5307T.
- ↑ "Library QC for ATAC-Seq and CUT&Tag". https://www.activemotif.com/blog-library-qc.
- ↑ 25.0 25.1 "High-throughput chromatin accessibility profiling at single-cell resolution". Nature Communications 9 (1). September 2018. doi:10.1038/s41467-018-05887-x. PMID 30194434. Bibcode: 2018NatCo...9.3647M.
- ↑ Cusanovich, Darren (May 2015). "Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing". Science 348 (6237): 910–914. doi:10.1126/science.aab1601. PMID 25953818. Bibcode: 2015Sci...348..910C.
- ↑ Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, Pokholok D, Aryee MJ, et al. (2019). "Droplet-based combinatorial indexing for massive scale single-cell epigenomics". bioRxiv 10.1101/612713.
- ↑ "A rapid and robust method for single cell chromatin accessibility profiling". Nature Communications 9 (1). December 2018. doi:10.1038/s41467-018-07771-0. PMID 30559361. Bibcode: 2018NatCo...9.5345C.
- ↑ Cao, Junyue; Cusanovich, Darren A.; Ramani, Vijay; Aghamirzaie, Delasa; Pliner, Hannah A.; Hill, Andrew J.; Daza, Riza M.; McFaline-Figueroa, Jose L. et al. (2018-09-28). "Joint profiling of chromatin accessibility and gene expression in thousands of single cells" (in en). Science 361 (6409): 1380–1385. doi:10.1126/science.aau0730. ISSN 0036-8075. PMID 30166440. Bibcode: 2018Sci...361.1380C.
- ↑ 30.0 30.1 Li, Zhijian; Kuppe, Christoph; Cheng, Mingbo; Menzel, Sylvia; Zenke, Martin; Kramann, Rafael et al. (2021). "Chromatin-accessibility estimation from single-cell ATAC-seq data with scOpen" (in en). Nature Communications 12 (1): 865931. doi:10.1038/s41467-021-26530-2. PMID 34737275. Bibcode: 2021NatCo..12.6386L.
- ↑ "chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data". Nature Methods 14 (10): 975–978. October 2017. doi:10.1038/nmeth.4401. PMID 28825706.
- ↑ 32.0 32.1 32.2 Zhang, Di; Deng, Yanxiang; Kukanja, Petra; Agirre, Eneritz; Bartosovic, Marek; Dong, Mingze; Ma, Cong; Ma, Sai et al. (April 2023). "Spatial epigenome-transcriptome co-profiling of mammalian tissues". Nature 616 (7955): 113–122. doi:10.1038/s41586-023-05795-1. ISSN 1476-4687. PMID 36922587. Bibcode: 2023Natur.616..113Z.
- ↑ "Spatial ATAC-Seq - CD Genomics". https://www.spatial-omicslab.com/spatial-atac-seq.html.
- ↑ Huang, Yung-Hsin; Belk, Julia A.; Zhang, Ruochi; Weiser, Natasha E.; Chiang, Zachary; Jones, Matthew G.; Mischel, Paul S.; Buenrostro, Jason D. et al. (2025-04-22). "Unified molecular approach for spatial epigenome, transcriptome, and cell lineages". Proceedings of the National Academy of Sciences 122 (16). doi:10.1073/pnas.2424070122. PMID 40249782. Bibcode: 2025PNAS..12224070H.
- ↑ Carraro, Caterina; Bonaguro, Lorenzo; Srinivasa, Rachana; Uelft, Martina van; Isakzai, Victoria; Schulte-Schrepping, Jonas; Gambhir, Prerna; Elmzzahi, Tarek et al. (2023-10-23). "Chromatin accessibility profiling of targeted cell populations with laser capture microdissection coupled to ATAC-seq" (in English). Cell Reports Methods 3 (10). doi:10.1016/j.crmeth.2023.100598. ISSN 2667-2375. PMID 37776856.
- ↑ Yuan, Ying; Ma, Cong; Yang, Mengnan (2020). "Spatially resolved multi-omics via deterministic barcoding in tissue". Cell 183 (6): 1665–1681.e18. doi:10.1016/j.cell.2020.10.026. PMID 33188776.
- ↑ Hamilton, Emily S. (2025). "Epigenomic landscape of the human dorsal root ganglion revealed by spatial ATAC-seq and CUT&Tag". PAIN 166 (3): 345–358. doi:10.1097/j.pain.0000000000003048. PMID 37831939.
- ↑ "Spatial chromatin accessibility mapping using deterministic barcoding". 2025. bioRxiv 10.1101/2025.02.25.638325.
- ↑ "Integrated spatial epigenomic profiling with DBiT-based ATAC-seq". 2025. bioRxiv 10.1101/2025.05.01.651759.
- ↑ Atkinson, Stuart P. (2022-10-10). "Spatial and Multimodal Updates to Single-cell ATAC-seq" (in en-US). https://epigenie.com/single-cell-spatial-and-multimodal-upgrades-to-atac-seq-speed-up-your-chromatin-insights/.
- ↑ Chang, Zhanhe; Xu, Yunfan; Dong, Xin; Gao, Yawei; Wang, Chenfei (2024-07-01). Nikolski, Macha. ed. "Single-cell and spatial multiomic inference of gene regulatory networks using SCRIPro" (in en). Bioinformatics 40 (7). doi:10.1093/bioinformatics/btae466. ISSN 1367-4811. PMID 39024032.
- ↑ Yan, Feng; Powell, David R.; Curtis, David J.; Wong, Nicholas C. (2020-02-03). "From reads to insight: a hitchhiker's guide to ATAC-seq data analysis". Genome Biology 21 (1): 22. doi:10.1186/s13059-020-1929-3. ISSN 1474-760X. PMID 32014034.
- ↑ Long, Yahui; Ang, Kok Siong; Sethi, Raman; Liao, Sha; Heng, Yang; van Olst, Lynn; Ye, Shuchen; Zhong, Chengwei et al. (September 2024). "Deciphering spatial domains from spatial multi-omics with SpatialGlue" (in en). Nature Methods 21 (9): 1658–1667. doi:10.1038/s41592-024-02316-4. ISSN 1548-7105. PMID 38907114.
- ↑ Zhang, Yong; Liu, Tao; Meyer, Clifford A.; Eeckhoute, Jérôme; Johnson, David S.; Bernstein, Bradley E.; Nusbaum, Chad; Myers, Richard M. et al. (2008-09-17). "Model-based Analysis of ChIP-Seq (MACS)". Genome Biology 9 (9): R137. doi:10.1186/gb-2008-9-9-r137. ISSN 1474-760X. PMID 18798982.
- ↑ Veerappa, Avinash M.; Rowley, M. Jordan; Maggio, Angela; Beaudry, Laura; Hawkins, Dale; Kim, Allen; Sethi, Sahil; Sorgen, Paul L. et al. (2024-07-23). "CloudATAC: a cloud-based framework for ATAC-Seq data analysis". Briefings in Bioinformatics 25 (Supplement_1). doi:10.1093/bib/bbae090. ISSN 1477-4054. PMID 39041910.
- ↑ Ewels, Philip A.; Peltzer, Alexander; Fillinger, Sven; Patel, Harshil; Alneberg, Johannes; Wilm, Andreas; Garcia, Maxime Ulysse; Di Tommaso, Paolo et al. (March 2020). "The nf-core framework for community-curated bioinformatics pipelines" (in en). Nature Biotechnology 38 (3): 276–278. doi:10.1038/s41587-020-0439-x. ISSN 1546-1696. https://www.nature.com/articles/s41587-020-0439-x.
- ↑ Chen, Huidong; Lareau, Caleb; Andreani, Tommaso; Vinyard, Michael E.; Garcia, Sara P.; Clement, Kendell; Andrade-Navarro, Miguel A.; Buenrostro, Jason D. et al. (2019-11-18). "Assessment of computational methods for the analysis of single-cell ATAC-seq data". Genome Biology 20 (1): 241. doi:10.1186/s13059-019-1854-5. ISSN 1474-760X. PMID 31739806.
External links
- ATAC-seq probes open-chromatin state (figure)
- ATAC-seq: Fast and sensitive epigenomic profiling
- HINT-ATAC: Identification of Transcription Factor Binding Sites using ATAC-seq
 |  |

