Biology:Isogenic human disease models
Isogenic human disease models are a family of cells that are selected or engineered to accurately model the genetics of a specific patient population, in vitro. They are provided with a genetically matched 'normal cell' to provide an isogenic system to research disease biology and novel therapeutic agents.[1] They can be used to model any disease with a genetic foundation. Cancer is one such disease for which isogenic human disease models have been widely used.
Historical models
Human isogenic disease models have been likened to 'patients in a test-tube', since they incorporate the latest research into human genetic diseases and do so without the difficulties and limitations involved in using non-human models.[2]
Historically, cells obtained from animals, typically mice, have been used to model cancer-related pathways. However, there are obvious limitations inherent in using animals for modelling genetically determined diseases in humans. Despite a large proportion of genetic conservation between humans and mice, there are significant differences between the biology of mice and humans that are important to cancer research. For example, major differences in telomere regulation enable murine cells to bypass the requirement for telomerase upregulation, which is a rate-limiting step in human cancer formation. As another example, certain ligand-receptor interactions are incompatible between mice and humans. Additionally, experiments have demonstrated important and significant differences in the ability to transform cells, compared with cells of murine origin. For these reasons, it remains essential to develop models of cancer that employ human cells.[3]
Targeting vectors
Isogenic cell lines are created via a process called homologous gene-targeting. Targeting vectors that utilize homologous recombination are the tools or techniques that are used to knock-in or knock-out the desired disease-causing mutation or SNP (single nucleotide polymorphism) to be studied. Although disease mutations can be harvested directly from cancer patients, these cells usually contain many background mutations in addition to the specific mutation of interest, and a matched normal cell line is typically not obtained. Subsequently, targeting vectors are used to 'knock-in' or 'knock out' gene mutations enabling a switch in both directions; from a normal to cancer genotype; or vice versa; in characterized human cancer cell lines such as HCT116 or Nalm6.[4]
There are several gene targeting technologies used to engineer the desired mutation, the most prevalent of which are briefly described, including key advantages and limitations, in the summary table below.
| Technique | Gene Knock-In | Gene Knock-out |
|---|---|---|
| rAAV (recombinant adeno-associated virus vectors)[5] | Targeted insertions or modifications are created within endogenous genes; and so are subject to:
rAAV can introduce subtle point mutations, SNPs as well as small insertions with high efficiency. Moreover, many peer reviewed studies have shown that rAAV does not introduce any confounding off target genomic events.[citation needed] Appears to be the preferred method being adopted in academia, Biotech and Pharma on a precision versus time versus cost basis.[citation needed]| |
Gene knockouts are at the endogenous locus, and thus are definitive, stable and patient relevant. No confounding off-target effects are elicited at other genomic loci. It requires a 2- step process:
This process can therefore generate 3 genotypes (+/+; -/+ and -/-); enabling therefore the analysis of haplo-insufficient gene function. Current limitation is the need to sequentially target single alleles making generation of knock-out cell lines a two-step process.| |
| Plasmid-based homologous recombination | Insertion is at the endogenous locus and has all the above benefits, but it is very inefficient. It also requires a promoterless drug selection strategy entailing bespoke construct generation. A large historical bank of cell lines has been generated using this method which has been displaced by other methods since the mid-1990s. | Deletion is at endogenous locus and has all the above benefits, but it is inefficient. It also requires a promoterless drug selection strategy that entails bespoke construct generation |
| Flip-in | This is an efficient technique that allows the directed insertion of 'ectopic' transgenes at a single pre-defined genomic locus (integration via a FLP recombinase site). This is not a technique for modifying an endogenous locus. Transgenes will usually be under the control of an exogenous promoter, or a partially defined promoter-unit in the incorrect genomic location. Their expression will therefore not be under the same genomic and epigenetic regulation as the endogenous loci, which limits the utility of these systems for studying gene-function. They are however, good for eliciting rapid and stable exogenous gene expression. | Not applicable |
| Zinc-Finger Nucleases (ZFNs) | ZFNs have been reported to achieve high rates of genetic knock-outs within a target endogenous gene. If ZFNs are co-delivered with a transgene construct homologous to the target gene, genetic knock-in's or insertions can also be achieved.[6] One potential drawback is that any off-target double strand breaks could lead to random off-target gene insertions, deletions and wider genomic instability; confounding the resulting genotype.[7] However, no measurable increase in the rate of random plasmid integration was observed in human cells efficiently edited with ZFNs that target a composite 24 bp recognition site [6] | ZFNs are sequence-directed endonucleases which enable the rapid and highly efficient (up to 90% in a bulk cell population) disruption of both alleles of a target gene, although user- defined or patient relevant loss of-function alterations have not been reported at similar frequencies. Off target deletions or insertions elsewhere in the genome are a significant concern. The speed advantage of obtaining a biallelic KO in one step is also partially mitigated if one still needs to derive a clonal cell line to study gene function in a homogenous cell-population. |
| Meganucleases | Meganucleases are operationally analogous to ZFN's. There are limitations inherent in their use such as the meganuclease vector design which can take up to 9 months and cost tens of thousands of dollars.[citation needed] This makes meganucleases more attractive in high-value applications such as gene therapy, agrobiotechnology and engineering of bioproducer lines. |
Homologous recombination in cancer cell disease models
Homologous recombination (HR) is a kind of genetic recombination in which genetic sequences are exchanged between two similar segments of DNA. HR plays a major role in eukaryotic cell division, promoting genetic diversity through the exchange between corresponding segments of DNA to create new, and potentially beneficial combinations of genes.[citation needed]
HR performs a second vital role in DNA repair, enabling the repair of double-strand breaks in DNA which is a common occurrence during a cell's lifecycle. It is this process which is artificially triggered by the above technologies and bootstrapped in order to engender 'knock-ins' or 'knockouts' in specific genes 5, 7.
A recent key advance was discovered using AAV-homologous recombination vectors, which increases the low natural rates of HR in differentiated human cells when combined with gene-targeting vectors-sequences.[citation needed]
-
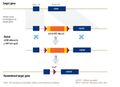 Diagram of a typical rAAV vector (source: https://www.horizondiscovery.com/gene-editing/raav)
Diagram of a typical rAAV vector (source: https://www.horizondiscovery.com/gene-editing/raav)

Commercialization
Factors leading to the recent commercialization of isogenic human cancer cell disease models for the pharmaceutical industry and research laboratories are twofold.[citation needed]
Firstly, successful patenting of enhanced targeting vector technology has provided a basis for commercialization of the cell-models which eventuate from the application of these technologies.[citation needed]
Secondly, the trend of relatively low success rates in pharmaceutical RnD and the enormous costs have created a real need for new research tools that illicit how patient sub-groups will respond positively or be resistant to targeted cancer therapeutics based upon their individual genetic profile.[citation needed]
See also
- AAV
- FLP-FRT recombination
- Genome engineering
- Homologous recombination
- Plasmid
- Recombinant AAV mediated genome engineering
- Synthetic lethality
- Zinc finger nuclease
References
- ↑ "Use of isogenic human cancer cells for high-throughput screening and drug discovery". Nat. Biotechnol. 19 (10): 940–5. October 2001. doi:10.1038/nbt1001-940. PMID 11581659.
- ↑ Gupta, Piyush B.; Kuperwasser, Charlotte (2004). "Disease models of breast cancer". Drug Discovery Today 1: 9–16. doi:10.1016/j.ddmod.2004.05.001.
- ↑ "Targeted transgene insertion into human chromosomes by adeno-associated virus vectors". Nat. Biotechnol. 20 (7): 735–8. July 2002. doi:10.1038/nbt0702-735. PMID 12089561.
- ↑ Masters JR (December 2000). "Human cancer cell lines: fact and fantasy". Nat. Rev. Mol. Cell Biol. 1 (3): 233–6. doi:10.1038/35043102. PMID 11252900.
- ↑ Engelhardt JF (August 2006). "AAV hits the genomic bull's-eye". Nat. Biotechnol. 24 (8): 949–50. doi:10.1038/nbt0806-949. PMID 16900138.
- ↑ 6.0 6.1 Urnov, Fyodor D.; Rebar, Edward J.; Holmes, Michael C.; Zhang, H. Steve; Gregory, Philip D. (2010). "Genome editing with engineered zinc finger nucleases". Nature Reviews Genetics 11 (9): 636–646. doi:10.1038/nrg2842. PMID 20717154.
- ↑ "Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications". Mol. Ther. 18 (4): 743–53. April 2010. doi:10.1038/mt.2009.304. PMID 20068556.
Sources
- "Mutational analysis of the tyrosine kinome in colorectal cancers". Science 300 (5621): 949. May 2003. doi:10.1126/science.1082596. PMID 12738854.
- "Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses". Nucleic Acids Res. 32 (1): 3e–3. 2004. doi:10.1093/nar/gnh009. PMID 14704360.
- "Mutational analysis of the tyrosine phosphatome in colorectal cancers". Science 304 (5674): 1164–6. May 2004. doi:10.1126/science.1096096. PMID 15155950. Bibcode: 2004Sci...304.1164W.
- "Improved methods for the generation of human gene knockout and knockin cell lines". Nucleic Acids Res. 33 (18): e158. 2005. doi:10.1093/nar/gni160. PMID 16214806.
- "Somatic mutation of EGFR catalytic domain and treatment with gefitinib in colorectal cancer". Ann. Oncol. 16 (11): 1848–9. November 2005. doi:10.1093/annonc/mdi356. PMID 16012179.
- "Kinase mutations in cancer: chinks in the enemy's armour?". Curr Opin Oncol 18 (1): 69–76. January 2006. doi:10.1097/01.cco.0000198020.91724.48. PMID 16357567.
- "Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies". Cancer Res. 67 (6): 2643–8. March 2007. doi:10.1158/0008-5472.CAN-06-4158. PMID 17363584. http://cancerres.aacrjournals.org/cgi/pmidlookup?view=long&pmid=17363584.
- "Genetic targeting of the kinase activity of the Met receptor in cancer cells". Proc. Natl. Acad. Sci. U.S.A. 104 (27): 11412–7. July 2007. doi:10.1073/pnas.0703205104. PMID 17595299. Bibcode: 2007PNAS..10411412A.
- "Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation". Cancer Res. 67 (18): 8460–7. September 2007. doi:10.1158/0008-5472.CAN-07-0108. PMID 17875684. http://cancerres.aacrjournals.org/cgi/pmidlookup?view=long&pmid=17875684.
- "Knock-in of oncogenic Kras does not transform mouse somatic cells but triggers a transcriptional response that classifies human cancers". Cancer Res. 67 (18): 8468–76. September 2007. doi:10.1158/0008-5472.CAN-07-1126. PMID 17875685. http://cancerres.aacrjournals.org/cgi/pmidlookup?view=long&pmid=17875685.
- "Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase". J. Cell Biol. 181 (6): 913–20. June 2008. doi:10.1083/jcb.200802076. PMID 18559665.
- "Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells". Proc. Natl. Acad. Sci. U.S.A. 105 (25): 8703–8. June 2008. doi:10.1073/pnas.0712060105. PMID 18562296. Bibcode: 2008PNAS..105.8703F.
- "Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer". J. Clin. Oncol. 26 (35): 5705–12. December 2008. doi:10.1200/JCO.2008.18.0786. PMID 19001320. http://www.jco.org/cgi/pmidlookup?view=long&pmid=19001320.
- "Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses". Proc. Natl. Acad. Sci. U.S.A. 105 (52): 20864–9. December 2008. doi:10.1073/pnas.0808757105. PMID 19106301. Bibcode: 2008PNAS..10520864D.
- "Knockin of mutant PIK3CA activates multiple oncogenic pathways". Proc. Natl. Acad. Sci. U.S.A. 106 (8): 2835–40. February 2009. doi:10.1073/pnas.0813351106. PMID 19196980. Bibcode: 2009PNAS..106.2835G.
- "PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies". Cancer Res. 69 (5): 1851–7. March 2009. doi:10.1158/0008-5472.CAN-08-2466. PMID 19223544. http://cancerres.aacrjournals.org/cgi/pmidlookup?view=long&pmid=19223544.
- "A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53". Proc. Natl. Acad. Sci. U.S.A. 106 (10): 3964–9. March 2009. doi:10.1073/pnas.0813333106. PMID 19225112. Bibcode: 2009PNAS..106.3964S.
- "Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells". Science 325 (5947): 1555–9. September 2009. doi:10.1126/science.1174229. PMID 19661383. Bibcode: 2009Sci...325.1555Y.
- Cordes, Nils, ed (2009). "Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer". PLOS ONE 4 (10): e7287. doi:10.1371/journal.pone.0007287. PMID 19806185. Bibcode: 2009PLoSO...4.7287S.
- Endogenous Expression of Oncogenic PI3K Mutation Leads to Activated PI3K Signaling and an Invasive Phenotype Poster Presented at AACR/EORTC Molecular Targets and Cancer Therapeutics, Boston, USA, Nov. 2009
- "Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer". J. Clin. Oncol. 28 (7): 1254–61. March 2010. doi:10.1200/JCO.2009.24.6116. PMID 20100961. http://www.jco.org/cgi/pmidlookup?view=long&pmid=20100961.
- Pearson, Christopher E., ed (February 2010). "Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells". PLOS Genet. 6 (2): e1000855. doi:10.1371/journal.pgen.1000855. PMID 20195511.
- Aziz, Syed A., ed (2010). "Use of human cancer cell lines mitochondria to explore the mechanisms of BH3 peptides and ABT-737-induced mitochondrial membrane permeabilization". PLOS ONE 5 (3): e9924. doi:10.1371/journal.pone.0009924. PMID 20360986. Bibcode: 2010PLoSO...5.9924B.
- Endogenous Expression of Oncogenic PI3K Mutation Leads to accumulation of anti-apoptotic proteins in mitochondria Poster Presented at AACR 2010, Washington, D.C., USA, April. 2010
- The use of 'X-MAN' isogenic cell lines to define PI3-kinase inhibitor activity profiles Poster Presented at AACR 2010, Washington, D.C., USA, April. 2010
- The use of 'X-MAN' mutant PI3CA increases the expression of individual tubulin isoforms and promoted resistance to anti-mitotic chemotherapy drugs Poster Presented at AACR 2010, Washington, D.C., USA, April. 2010
- "Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus". J. Clin. Invest. 120 (8): 2858–66. August 2010. doi:10.1172/JCI37539. PMID 20664172.
 |  |

