Software:TURBOMOLE
 | |
| Developer(s) | Turbomole GmbH |
|---|---|
| Stable release | TURBOMOLE 7.6
|
| Operating system | Linux, Windows, Mac OS |
| Type | Computational Chemistry |
| License | Commercial |
| Website | www |
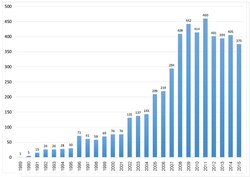
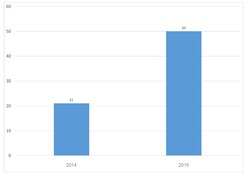
TURBOMOLE is an ab initio computational chemistry program that implements various quantum chemistry methods. It was initially developed by the group of Prof. Reinhart Ahlrichs at the University of Karlsruhe. In 2007, TURBOMOLE GmbH, founded by R. Ahlrichs, F. Furche, C. Hättig, W. Klopper, M. Sierka, and F. Weigend, took over the responsibility for the coordination of the scientific development of TURBOMOLE program, for which the company holds all copy and intellectual property rights. In 2018 David P. Tew joined the TURBOMOLE GmbH. Since 1987, this program is one of the useful tools as it involves in many fields of research including heterogeneous and homogeneous catalysis, organic and inorganic chemistry, spectroscopy as well as biochemistry. This can be illustrated by citation records of Ahlrich's 1989 publication which is more than 6700 times as of 18 July 2020.[1] In the year 2014,[2] the second Turbomole article has been published. The number of citations from both papers indicates that the Turbomole's user base is expanding.
General features
Turbomole was developed in 1987 and turned into a mature program system under the control of Reinhart Ahlrichs and his collaborators. Turbomole can perform a large-scale quantum chemical simulations of molecules, clusters, and later periodic solids. Gaussian basis sets are used in Turbomole. The functionality of the program concentrates extensively on the electronic structure methods with effective cost-performance characteristics such as density functional theory,[3] second–order Møller-Plesset[4][5] and coupled cluster theory. Aside from energies and structures, an assortment of optical, electrical, and magnetic properties are available from analytical energy derivative for electronic ground and excited states.[2] However, up to the year 2000, Turbomole was only limited to the calculation of molecules in gas phase, thus, COSMO has been implemented in the Turbomole in a cooperative initiative of BASF AG and Bayer AG.[6] Turbomole version 6.5 releasing in the year 2013, comes with post-Kohn–Sham calculations within the random-phase approximation. Turbomole also comes with another significant additions including nonadiabatic molecular dynamics, ultra-efficient higher order CC methods, new density functionals and periodic calculations.[7] TmoleX is available as a graphical user interface for Turbomole allowing the user to perform the entire workflow of a quantum chemical investigation ranging from building of an initial structure to the interpretation of the results.[8]
Version history
The current version of Turbomole is V7.8 released in December 2023[7]
References
- ↑ Ahlrichs, Reinhart; Bär, Michael; Häser, Marco; Hom, Hans; Kölmel, Christoph (1989). "Electronic structure calculations on workstation computers". Chemical Physics Letters 162 (3): 165–169. doi:10.1016/0009-2614(89)85118-8. Bibcode: 1989CPL...162..165A.
- ↑ 2.0 2.1 Furche, Filipp; Ahlrichs, Reinhart; Hättig, Christof; Klopper, Wim; Sierka, Marek; Weigend, Florian (2014). "Turbomole". WIREs Comput Mol Sci 4 (2): 91–100. doi:10.1002/wcms.1162.
- ↑ Ahlrichs, Reinhart; Arnim, Malte V. (1998). "Performance of parallel Turbomole for density functional calculations". Journal of Computational Chemistry 19 (15): 1746–1757. doi:10.1002/(SICI)1096-987X(19981130)19:15<1746::AID-JCC7>3.0.CO;2-N.
- ↑ Bachorz, Rafal A.; Bischoff, Florian A.; Glöb, Andreas; Hättig, Christof; Klopper, Wim; Tew, David P. (2011). "Software news and update: The MP2-F12 method in the Turbomole program package". Journal of Computational Chemistry 32 (11): 2492–2513. doi:10.1002/jcc.21825. PMID 21590779.
- ↑ Gerenkamp, Mareike; Grimme, Stefan (2004). "Spin-component scaled second-order Møller-Plesset perturbation theory for the calculation of molecular geometries and harmonic vibrational frequencies". Chemical Physics Letters 392 (1–3): 229–235. doi:10.1016/j.cplett.2004.05.063. Bibcode: 2004CPL...392..229G.
- ↑ Schäfer, Ansgar; Klamt, Andreas; Sattel, Diana; Lohrenz, John C.W.; Eckert, Frank (2000). "COSMO implementation in Turbomole: Extension of an efficient quantum chemical code towards liquid systems". Physical Chemistry Chemical Physics 2 (10): 2187–2193. doi:10.1039/B000184H. Bibcode: 2000PCCP....2.2187S. https://zenodo.org/record/1452454.
- ↑ 7.0 7.1 "Turbomole Release Note". Cosmologic. http://www.cosmologic.de/turbomole/product-info/release-notes.html.
- ↑ Steffen, Claudia; Thomas, Klaus; Huniar, Uwe; Hellweg, Arnim; Rubner, Oliver; Schroer, Alexander (2010). "Software news and update: TmoleX-A graphical user interface for Turbomole". Journal of Computational Chemistry 31 (16): 2967–2970. doi:10.1002/jcc.21576. PMID 20928852.
External links
 |  |

