Chemistry:Heterogeneous catalysis

Heterogeneous catalysis is catalysis where the phase of catalysts differs from that of the reagents or products.[1] The process contrasts with homogeneous catalysis where the reagents, products and catalyst exist in the same phase. Phase distinguishes between not only solid, liquid, and gas components, but also immiscible mixtures (e.g., oil and water), or anywhere an interface is present.
Heterogeneous catalysis typically involves solid phase catalysts and gas phase reactants.[2] In this case, there is a cycle of molecular adsorption, reaction, and desorption occurring at the catalyst surface. Thermodynamics, mass transfer, and heat transfer influence the rate (kinetics) of reaction.
Heterogeneous catalysis is very important because it enables faster, large-scale production and the selective product formation.[3] Approximately 35% of the world's GDP is influenced by catalysis.[4] The production of 90% of chemicals (by volume) is assisted by solid catalysts.[2] The chemical and energy industries rely heavily on heterogeneous catalysis. For example, the Haber–Bosch process uses metal-based catalysts in the synthesis of ammonia, an important component in fertilizer; 144 million tons of ammonia were produced in 2016.[5]
Adsorption
Adsorption is an essential step in heterogeneous catalysis. Adsorption is the process by which a gas (or solution) phase molecule (the adsorbate) binds to solid (or liquid) surface atoms (the adsorbent). The reverse of adsorption is desorption, the adsorbate splitting from adsorbent. In a reaction facilitated by heterogeneous catalysis, the catalyst is the adsorbent and the reactants are the adsorbate.
Types of adsorption
Two types of adsorption are recognized: physisorption, weakly bound adsorption, and chemisorption, strongly bound adsorption. Many processes in heterogeneous catalysis lie between the two extremes. The Lennard-Jones model provides a basic framework for predicting molecular interactions as a function of atomic separation.[6]
Physisorption
In physisorption, a molecule becomes attracted to the surface atoms via van der Waals forces. These include dipole-dipole interactions, induced dipole interactions, and London dispersion forces. Note that no chemical bonds are formed between adsorbate and adsorbent, and their electronic states remain relatively unperturbed. Typical energies for physisorption are from 3 to 10 kcal/mol.[2] In heterogeneous catalysis, when a reactant molecule physisorbs to a catalyst, it is commonly said to be in a precursor state, an intermediate energy state before chemisorption, a more strongly bound adsorption.[6] From the precursor state, a molecule can either undergo chemisorption, desorption, or migration across the surface.[7] The nature of the precursor state can influence the reaction kinetics.[7]
Chemisorption
When a molecule approaches close enough to surface atoms such that their electron clouds overlap, chemisorption can occur. In chemisorption, the adsorbate and adsorbent share electrons signifying the formation of chemical bonds. Typical energies for chemisorption range from 20 to 100 kcal/mol.[2] Two cases of chemisorption are:
- Molecular adsorption: the adsorbate remains intact. An example is alkene binding by platinum.
- Dissociation adsorption: one or more bonds break concomitantly with adsorption. In this case, the barrier to dissociation affects the rate of adsorption. An example of this is the binding of H2 to a metal catalyst, where the H-H bond is broken upon adsorption.
Surface reactions

Most metal surface reactions occur by chain propagation in which catalytic intermediates are cyclically produced and consumed.[8] Two main mechanisms for surface reactions can be described for A + B → C.[2]
- Langmuir–Hinshelwood mechanism: The reactant molecules, A and B, both adsorb to the catalytic surface. While adsorbed to the surface, they combine to form product C, which then desorbs.
- Eley–Rideal mechanism: One reactant molecule, A, adsorbs to the catalytic surface. Without adsorbing, B reacts with absorbed A to form C, that then desorbs from the surface.
Most heterogeneously catalyzed reactions are described by the Langmuir–Hinshelwood model.[9]
In heterogeneous catalysis, reactants diffuse from the bulk fluid phase to adsorb to the catalyst surface. The adsorption site is not always an active catalyst site, so reactant molecules must migrate across the surface to an active site. At the active site, reactant molecules will react to form product molecule(s) by following a more energetically facile path through catalytic intermediates (see figure to the right). The product molecules then desorb from the surface and diffuse away. The catalyst itself remains intact and free to mediate further reactions. Transport phenomena such as heat and mass transfer, also play a role in the observed reaction rate.
Catalyst design

Catalysts are not active towards reactants across their entire surface; only specific locations possess catalytic activity, called active sites. The surface area of a solid catalyst has a strong influence on the number of available active sites. In industrial practice, solid catalysts are often porous to maximize surface area, commonly achieving 50–400 m2/g.[2] Some mesoporous silicates, such as the MCM-41, have surface areas greater than 1000 m2/g.[10] Porous materials are cost effective due to their high surface area-to-mass ratio and enhanced catalytic activity.
In many cases, a solid catalyst is dispersed on a supporting material to increase surface area (spread the number of active sites) and provide stability.[2] Usually catalyst supports are inert, high melting point materials, but they can also be catalytic themselves. Most catalyst supports are porous (frequently carbon, silica, zeolite, or alumina-based)[4] and chosen for their high surface area-to-mass ratio. For a given reaction, porous supports must be selected such that reactants and products can enter and exit the material.
Often, substances are intentionally added to the reaction feed or on the catalyst to influence catalytic activity, selectivity, and/or stability. These compounds are called promoters. For example, alumina (Al2O3) is added during ammonia synthesis to providing greater stability by slowing sintering processes on the Fe-catalyst.[2]
Sabatier principle can be considered one of the cornerstones of modern theory of catalysis.[11] Sabatier principle states that the surface-adsorbates interaction has to be an optimal amount: not too weak to be inert toward the reactants and not too strong to poison the surface and avoid desorption of the products.[12] The statement that the surface-adsorbate interaction has to be an optimum, is a qualitative one. Usually the number of adsorbates and transition states associated with a chemical reaction is a large number, thus the optimum has to be found in a many-dimensional space. Catalyst design in such a many-dimensional space is not a computationally viable task. Additionally, such optimization process would be far from intuitive. Scaling relations are used to decrease the dimensionality of the space of catalyst design.[13] Such relations are correlations among adsorbates binding energies (or among adsorbate binding energies and transition states also known as BEP relations)[14] that are "similar enough" e.g., OH versus OOH scaling.[15] Applying scaling relations to the catalyst design problems greatly reduces the space dimensionality (sometimes to as small as 1 or 2).[16] One can also use micro-kinetic modeling based on such scaling relations to take into account the kinetics associated with adsorption, reaction and desorption of molecules under specific pressure or temperature conditions.[17] Such modeling then leads to well-known volcano-plots at which the optimum qualitatively described by the Sabatier principle is referred to as the "top of the volcano". Scaling relations can be used not only to connect the energetics of radical surface-adsorbed groups (e.g., O*,OH*),[13] but also to connect the energetics of closed-shell molecules among each other or to the counterpart radical adsorbates.[18] A recent challenge for researchers in catalytic sciences is to "break" the scaling relations.[19] The correlations which are manifested in the scaling relations confine the catalyst design space, preventing one from reaching the "top of the volcano". Breaking scaling relations can refer to either designing surfaces or motifs that do not follow a scaling relation, or ones that follow a different scaling relation (than the usual relation for the associated adsorbates) in the right direction: one that can get us closer to the top of the reactivity volcano.[16] In addition to studying catalytic reactivity, scaling relations can be used to study and screen materials for selectivity toward a special product.[20] There are special combination of binding energies that favor specific products over the others. Sometimes a set of binding energies that can change the selectivity toward a specific product "scale" with each other, thus to improve the selectivity one has to break some scaling relations; an example of this is the scaling between methane and methanol oxidative activation energies that leads to the lack of selectivity in direct conversion of methane to methanol.[21]
Catalyst deactivation
Catalyst deactivation is defined as a loss in catalytic activity and/or selectivity over time.
Substances that decrease the reaction rate are called poisons. Poisons chemisorb to the catalyst surface and reduce the number of available active sites for reactant molecules to bind to.[22] Common poisons include Group V, VI, and VII elements (e.g. S, O, P, Cl), some toxic metals (e.g. As, Pb), and adsorbing species with multiple bonds (e.g. CO, unsaturated hydrocarbons).[6][22] For example, sulfur disrupts the production of methanol by poisoning the Cu/ZnO catalyst.[23] Substances that increase reaction rate are called promoters. For example, the presence of alkali metals in ammonia synthesis increases the rate of N2 dissociation.[23]
The presence of poisons and promoters can alter the activation energy of the rate-limiting step and affect a catalyst's selectivity for the formation of certain products. Depending on the amount, a substance can be favorable or unfavorable for a chemical process. For example, in the production of ethylene, a small amount of chemisorbed chlorine will act as a promoter by improving Ag-catalyst selectivity towards ethylene over CO2, while too much chlorine will act as a poison.[6]
Other mechanisms for catalyst deactivation include:
- Sintering: when heated, dispersed catalytic metal particles can migrate across the support surface and form crystals. This results in a reduction of catalyst surface area.
- Fouling: the deposition of materials from the fluid phase onto the solid phase catalyst and/or support surfaces. This results in active site and/or pore blockage.
- Coking: the deposition of heavy, carbon-rich solids onto surfaces due to the decomposition of hydrocarbons[22]
- Vapor-solid reactions: formation of an inactive surface layer and/or formation of a volatile compound that exits the reactor.[22] This results in a loss of surface area and/or catalyst material.
- Solid-state transformation: solid-state diffusion of catalyst support atoms to the surface followed by a reaction that forms an inactive phase. This results in a loss of catalyst surface area.
- Erosion: continual attrition of catalyst material common in fluidized-bed reactors.[24] This results in a loss of catalyst material.
In industry, catalyst deactivation costs billions every year due to process shutdown and catalyst replacement.[22]
Industrial examples
In industry, many design variables must be considered including reactor and catalyst design across multiple scales ranging from the subnanometer to tens of meters. The conventional heterogeneous catalysis reactors include batch, continuous, and fluidized-bed reactors, while more recent setups include fixed-bed, microchannel, and multi-functional reactors.[6] Other variables to consider are reactor dimensions, surface area, catalyst type, catalyst support, as well as reactor operating conditions such as temperature, pressure, and reactant concentrations.

Some large-scale industrial processes incorporating heterogeneous catalysts are listed below.[4]
| Process | Reactants, Product/s (not balanced) | Catalyst | Comment |
|---|---|---|---|
| Sulfuric acid synthesis (Contact process) | SO2 + O2, SO3 | vanadium oxides | Hydration of SO3 gives H2SO4 |
| Ammonia synthesis (Haber–Bosch process) | N2 + H2, NH3 | iron oxides on alumina(Al2O3) | Consumes 1% of world's industrial energy budget[2] |
| Nitric acid synthesis (Ostwald process) | NH3 + O2, HNO3 | unsupported Pt-Rh gauze | Direct routes from N2 are uneconomical |
| Hydrogen production by Steam reforming | CH4 + H2O, H2 + CO2 | Nickel or K2O | Greener routes to H2 by water splitting actively sought |
| Ethylene oxide synthesis | C2H4 + O2, C2H4O | silver on alumina, with many promoters | Poorly applicable to other alkenes |
| Hydrogen cyanide synthesis (Andrussov oxidation) | NH3 + O2 + CH4, HCN | Pt-Rh | Related ammoxidation process converts hydrocarbons to nitriles |
| Olefin polymerization Ziegler–Natta polymerization | propylene, polypropylene | TiCl3 on MgCl2 | Many variations exist, including some homogeneous examples |
| Desulfurization of petroleum (hydrodesulfurization) | H2 + R2S (idealized organosulfur impurity), RH + H2S | Mo-Co on alumina | Produces low-sulfur hydrocarbons, sulfur recovered via the Claus process |

Other examples
- Reduction of nitriles in the synthesis of phenethylamine with Raney nickel catalyst and hydrogen in ammonia:[25]
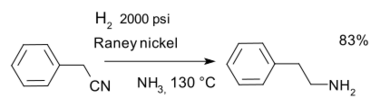
Nitrile hydrogenation - The cracking, isomerisation, and reformation of hydrocarbons to form appropriate and useful blends of petrol.
- In automobiles, catalytic converters are used to catalyze three main reactions:
- The oxidation of carbon monoxide to carbon dioxide:
- 2CO(g) + O2(g) → 2CO2(g)
- The reduction of nitrogen monoxide back to nitrogen:
- 2NO(g) + 2CO(g) → N2(g) + 2CO2(g)
- The oxidation of hydrocarbons to water and carbon dioxide:
- 2 C6H6 + 15 O2 → 12 CO2 + 6 H2O
- The oxidation of carbon monoxide to carbon dioxide:
- This process can occur with any of hydrocarbon, but most commonly is performed with petrol or diesel.
- Asymmetric heterogeneous catalysis facilitates the production of pure enantiomer compounds using chiral heterogeneous catalysts.[26]
- The majority of heterogeneous catalysts are based on metals[27] or metal oxides;[28] however, some chemical reactions can be catalyzed by carbon-based materials, e.g., oxidative dehydrogenations[29] or selective oxidations.[30]
- Ethylbenzene + 1/2 O2 → Styrene + H2O
- Acrolein + 1/2 O2 → Acrylic acid

Solid-Liquid and Liquid-Liquid Catalyzed Reactions
Although the majority of heterogeneous catalysts are solids, there are a few variations which are of practical value. For two immiscible solutions (liquids), one carries the catalyst while the other carries the reactant. This set up is the basis of biphasic catalysis as implemented in the industrial production of butyraldehyde by the hydroformylation of propylene.[31]
| Reacting phases | Examples given | Comment |
|---|---|---|
| solid + solution | hydrogenation of fatty acids with nickel | used for the production of margarine |
| immiscible liquid phases | hydroformylation of propene | aqueous phase catalyst; reactants and products mainly in non-aqueous phase |
See also
- Heterogeneous gold catalysis
- Nanomaterial-based catalysts
- Platinum nanoparticles
- Temperature-programmed reduction
- Thermal desorption spectroscopy
References
- ↑ Schlögl, Robert (9 March 2015). "Heterogeneous Catalysis". Angewandte Chemie International Edition 54 (11): 3465–3520. doi:10.1002/anie.201410738. PMID 25693734.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 Rothenberg, Gadi (17 March 2008). Catalysis: concepts and green applications. Weinheim [Germany]: Wiley-VCH. ISBN 978-3-527-31824-7. OCLC 213106542.
- ↑ Information., Lawrence Berkeley National Laboratory. United States. Department of Energy. Office of Scientific and Technical (2003). "The impact of nanoscience on heterogeneous catalysis". Science (Lawrence Berkeley National Laboratory) 299 (5613): 1688–1691. doi:10.1126/science.1083671. OCLC 727328504. PMID 12637733. Bibcode: 2003Sci...299.1688B. https://digital.library.unt.edu/ark:/67531/metadc780930/.
- ↑ 4.0 4.1 4.2 Ma, Zhen; Zaera, Francisco (2006-03-15), King, R. Bruce; Crabtree, Robert H.; Lukehart, Charles M. et al., eds., Heterogeneous Catalysis by Metals, John Wiley & Sons, Ltd, doi:10.1002/0470862106.ia084, ISBN 978-0-470-86078-6
- ↑ "United States Geological Survey, Mineral Commodity Summaries". January 2018. https://minerals.usgs.gov/minerals/pubs/commodity/nitrogen/mcs-2018-nitro.pdf.
- ↑ 6.0 6.1 6.2 6.3 6.4 Thomas, J. M.; Thomas, W. J. (2014-11-19). Principles and practice of heterogeneous catalysis (Second, revised ed.). Weinheim, Germany. ISBN 978-3-527-68378-9. OCLC 898421752.
- ↑ 7.0 7.1 Bowker, Michael (2016-03-28). "The Role of Precursor States in Adsorption, Surface Reactions and Catalysis". Topics in Catalysis 59 (8–9): 663–670. doi:10.1007/s11244-016-0538-6. ISSN 1022-5528. PMID 21386456. https://orca.cardiff.ac.uk/id/eprint/91556/1/art_10.1007_s11244-016-0538-6.pdf.
- ↑ Masel, Richard I. (22 March 1996) (in en). Principles of Adsorption and Reaction on Solid Surfaces. Wiley. ISBN 978-0-471-30392-3. OCLC 32429536. https://books.google.com/books?id=kEpRAAAAMAAJ.
- ↑ Petukhov, A.V. (1997). "Effect of molecular mobility on kinetics of an electrochemical Langmuir–Hinshelwood reaction". Chemical Physics Letters 277 (5–6): 539–544. doi:10.1016/s0009-2614(97)00916-0. ISSN 0009-2614. Bibcode: 1997CPL...277..539P.
- ↑ Kresge, C. T.; Leonowicz, M. E.; Roth, W. J.; Vartuli, J. C.; Beck, J. S. (1992). "Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism". Nature 359 (6397): 710–712. doi:10.1038/359710a0. ISSN 0028-0836. Bibcode: 1992Natur.359..710K.
- ↑ Medford, Andrew J.; Vojvodic, Aleksandra; Hummelshøj, Jens S.; Voss, Johannes; Abild-Pedersen, Frank; Studt, Felix; Bligaard, Thomas; Nilsson, Anders et al. (2015). "From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis". Journal of Catalysis 328: 36–42. doi:10.1016/j.jcat.2014.12.033.
- ↑ Laursen, Anders B.; Man, Isabela Costinela; Trinhammer, Ole L.; Rossmeisl, Jan; Dahl, Søren (2011-10-04). "The Sabatier Principle Illustrated by Catalytic H2O2 Decomposition on Metal Surfaces". Journal of Chemical Education 88 (12): 1711–1715. doi:10.1021/ed101010x. Bibcode: 2011JChEd..88.1711L.
- ↑ 13.0 13.1 Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T. R.; Moses, P. G.; Skúlason, E.; Bligaard, T. et al. (2007-07-06). "Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces". Physical Review Letters 99 (1). doi:10.1103/PhysRevLett.99.016105. PMID 17678168. Bibcode: 2007PhRvL..99a6105A. http://orbit.dtu.dk/ws/files/4787818/Scaling_PRL.pdf.
- ↑ Nørskov, Jens K.; Christensen, Claus H.; Bligaard, Thomas; Munter, Ture R. (2008-08-18). "BEP relations for N2 dissociation over stepped transition metal and alloy surfaces". Physical Chemistry Chemical Physics 10 (34): 5202–5206. doi:10.1039/B720021H. ISSN 1463-9084. PMID 18728861. Bibcode: 2008PCCP...10.5202M.
- ↑ Viswanathan, Venkatasubramanian; Hansen, Heine Anton; Rossmeisl, Jan; Nørskov, Jens K. (2012-07-11). "Universality in Oxygen Reduction Electrocatalysis on Metal Surfaces". ACS Catalysis 2 (8): 1654–1660. doi:10.1021/cs300227s. ISSN 2155-5435.
- ↑ 16.0 16.1 Nørskov, Jens K.; Vojvodic, Aleksandra (2015-06-01). "New design paradigm for heterogeneous catalysts". National Science Review 2 (2): 140–143. doi:10.1093/nsr/nwv023. ISSN 2095-5138.
- ↑ Medford, Andrew J.; Shi, Chuan; Hoffmann, Max J.; Lausche, Adam C.; Fitzgibbon, Sean R.; Bligaard, Thomas; Nørskov, Jens K. (2015-03-01). "CatMAP: A Software Package for Descriptor-Based Microkinetic Mapping of Catalytic Trends". Catalysis Letters 145 (3): 794–807. doi:10.1007/s10562-015-1495-6. ISSN 1572-879X.
- ↑ Kakekhani, Arvin; Roling, Luke T.; Kulkarni, Ambarish; Latimer, Allegra A.; Abroshan, Hadi; Schumann, Julia; AlJama, Hassan; Siahrostami, Samira et al. (2018-06-18). "Nature of Lone-Pair–Surface Bonds and Their Scaling Relations". Inorganic Chemistry 57 (12): 7222–7238. doi:10.1021/acs.inorgchem.8b00902. ISSN 0020-1669. PMID 29863849.
- ↑ Chen, Ping; He, Teng; Wu, Guotao; Guo, Jianping; Gao, Wenbo; Chang, Fei; Wang, Peikun (January 2017). "Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation". Nature Chemistry 9 (1): 64–70. doi:10.1038/nchem.2595. ISSN 1755-4349. PMID 27995914. Bibcode: 2017NatCh...9...64W.
- ↑ Schumann, Julia; Medford, Andrew J.; Yoo, Jong Suk; Zhao, Zhi-Jian; Bothra, Pallavi; Cao, Ang; Studt, Felix; Abild-Pedersen, Frank et al. (2018-03-13). "Selectivity of Synthesis Gas Conversion to C2+ Oxygenates on fcc(111) Transition-Metal Surfaces". ACS Catalysis 8 (4): 3447–3453. doi:10.1021/acscatal.8b00201. https://www.osti.gov/biblio/1457170.
- ↑ Nørskov, Jens K.; Studt, Felix; Abild-Pedersen, Frank; Tsai, Charlie; Yoo, Jong Suk; Montoya, Joseph H.; Aljama, Hassan; Kulkarni, Ambarish R. et al. (February 2017). "Understanding trends in C–H bond activation in heterogeneous catalysis". Nature Materials 16 (2): 225–229. doi:10.1038/nmat4760. ISSN 1476-4660. PMID 27723737. Bibcode: 2017NatMa..16..225L. https://escholarship.org/uc/item/2ww8j3wc.
- ↑ 22.0 22.1 22.2 22.3 22.4 Bartholomew, Calvin H (2001). "Mechanisms of catalyst deactivation". Applied Catalysis A: General 212 (1–2): 17–60. doi:10.1016/S0926-860X(00)00843-7. Bibcode: 2001AppCA.212...17B.
- ↑ 23.0 23.1 Nørskov, Jens K. (2014-08-25). Fundamental concepts in heterogeneous catalysis. Studt, Felix., Abild-Pedersen, Frank., Bligaard, Thomas.. Hoboken, New Jersey. ISBN 978-1-118-89202-2. OCLC 884500509.
- ↑ Forzatti, P (1999-09-14). "Catalyst deactivation". Catalysis Today 52 (2–3): 165–181. doi:10.1016/s0920-5861(99)00074-7. ISSN 0920-5861.
- ↑ Organic Syntheses, Coll. Vol. 3, p.720 (1955); Vol. 23, p.71 (1943). https://web.archive.org/web/20120315000000*/http://orgsynth.org/orgsyn/pdfs/CV4P0603.pdf
- ↑ Heitbaum; Glorius; Escher (2006). "Asymmetric heterogeneous catalysis". Angew. Chem. Int. Ed. 45 (29): 4732–62. doi:10.1002/anie.200504212. PMID 16802397.
- ↑ Wang, Aiqin; Li, Jun; Zhang, Tao (June 2018). "Heterogeneous single-atom catalysis" (in en). Nature Reviews Chemistry 2 (6): 65–81. doi:10.1038/s41570-018-0010-1. ISSN 2397-3358.
- ↑ Zeng, Liang; Cheng, Zhuo; Fan, Jonathan A.; Fan, Liang-Shih; Gong, Jinlong (November 2018). "Metal oxide redox chemistry for chemical looping processes" (in en). Nature Reviews Chemistry 2 (11): 349–364. doi:10.1038/s41570-018-0046-2. ISSN 2397-3358.
- ↑ Zhang, J.; Liu, X.; Blume, R.; Zhang, A.; Schlögl, R.; Su, D. S. (2008). "Surface-Modified Carbon Nanotubes Catalyze Oxidative Dehydrogenation of n-Butane". Science 322 (5898): 73–77. doi:10.1126/science.1161916. PMID 18832641. Bibcode: 2008Sci...322...73Z.
- ↑ Frank, B.; Blume, R.; Rinaldi, A.; Trunschke, A.; Schlögl, R. (2011). "Oxygen Insertion Catalysis by sp2 Carbon". Angew. Chem. Int. Ed. 50 (43): 10226–10230. doi:10.1002/anie.201103340. PMID 22021211.
- ↑ Boy Cornils, ed (2004). Aqueous-Phase Organometallic Catalysis: Concepts and Applications. Wiley-VCH.
External links
 |  |

