Chemistry:Sulfur isotope biogeochemistry
Sulfur isotope biogeochemistry is the study of the distribution of sulfur isotopes in biological and geological materials. In addition to its common isotope, 32S, sulfur has three rare stable isotopes: 34S, 36S, and 33S. The distribution of these isotopes in the environment is controlled by many biochemical and physical processes, including biological metabolisms, mineral formation processes, and atmospheric chemistry. Measuring the abundance of sulfur stable isotopes in natural materials, like bacterial cultures, minerals, or seawater, can reveal information about these processes both in the modern environment and over Earth history.[1]
Background
Natural abundance of sulfur isotopes

Sulfur has 24 known isotopes,[2] 4 of which are stable (meaning that they do not undergo radioactive decay).[3] 32S, the common isotope of sulfur, makes up 95.0% of the natural sulfur on Earth.[2] In the atomic symbol of 32S, the number 32 refers to the mass of each sulfur atom in daltons, the result of the 16 protons and 16 neutrons of 1 dalton each that make up the sulfur nucleus. The three rare stable isotopes of sulfur are 34S (4.2% of natural sulfur), 33S (0.75%), and 36S (0.015%).[4] These isotopes differ from 32S in the number of neutrons in each atom, but not the number of protons or electrons; as a result, each isotope has a slightly different mass, but has nearly identical chemical properties.[3]
Physical chemistry
Small differences in mass between stable isotopes of the same element can lead to a phenomenon called an "isotope effect," where heavier or lighter isotopes are preferentially incorporated into different natural materials depending on the materials' chemical composition or physical state.[5] Isotope effects are divided into two main groups: kinetic isotope effects and equilibrium isotope effects.[5] A kinetic isotope effect occurs when a reaction is irreversible, meaning that the reaction only proceeds in the direction from reactants to products.[3][5] Kinetic isotope effects cause isotopic fractionation—meaning that they affect the isotopic composition of reactant and product compounds—because the mass differences between stable isotopes can affect the rate of chemical reactions.[5] It takes more energy to reach the transition state of a reaction if the compound has bonds with a heavier isotope, which causes the compound with heavier isotopes to react more slowly.[5] Normal kinetic isotope effects cause the lighter isotope (or isotopes) to be preferentially included in a reaction's product.[5] The products are then said to be "depleted" in the heavy isotope relative to the reactant.[3] Rarely, inverse kinetic isotope effects may occur, where the heavier isotope is preferentially included in a reaction's product.[5][6]
Equilibrium isotope effects cause fractionation because it is more chemically favorable for heavy isotopes to take part in stronger bonds.[5] An equilibrium isotope effect occurs when a reaction is at equilibrium, meaning that the reaction is able to occur in both directions simultaneously.[3] When a reaction is at equilibrium, heavy isotopes will preferentially accumulate where they can form the strongest bonds.[3] For example, when the water in a sealed, half-full bottle is in equilibrium with the vapor above it, the heavier isotopes 2H and 18O will accumulate in the liquid, where they form stronger bonds, while the lighter isotopes 1H and 16O will accumulate in the vapor.[7] The liquid is then said to be "enriched" in the heavy isotope relative to the vapor.[3]
Calculations
Delta notation
Differences in the abundance of stable isotopes among natural materials are usually very small (natural differences in the ratio of rare to common isotope are almost always below 0.1%, and sometimes much smaller).[5] Nevertheless, these very small differences can record meaningful biological and geological processes. To facilitate comparison of these small but meaningful differences, isotope abundances in natural materials are often reported relative to isotope abundances in designated standards.[3][5] The convention for reporting the measured difference between a sample and a standard is called "delta notation." For example, imagine an element X for which we wish to compare the rare, heavy stable isotope with atomic mass A (AX) to the light, common isotope with atomic mass B (BX). The abundance of AX and BX in any given material is reported with the notation δAX. δAX for the sample material is calculated as follows:[5]
- AR = (total amount of AX)/(total amount of BX)
- δAXsample = (ARsample − ARstandard)/ARstandard
δ values are most commonly reported in parts per thousand, commonly referred to in isotope chemistry as per mille and represented by the symbol ‰. To report δ values in per mille, the δ value as calculated above should be multiplied by 1000:
- δAXsample (‰) = ((ARsample − ARstandard)/ARstandard) * 1000
Fractionation factors
While an isotope effect is the physical tendency for stable isotopes to distribute in a particular way, the isotopic fractionation is the measurable result of this tendency.[5] The isotopic fractionation of a natural process can be calculated from measured isotope abundances. The calculated value is called a "fractionation factor," and allows the effect of different processes on isotope distributions to be mathematically compared.[5] For example, imagine a chemical reaction Reactant → Product. Reactant and Product are materials that both contain the element X, and X has two stable isotopes, AX (the heavy isotope, with a mass of A) and BX (the light isotope, with a mass of B). The fractionation factor for the element X in the reaction Reactant → Product is represented by the notation
- AαProduct/Reactant. AαProduct/Reactant is calculated as follows:[5]
- AαProduct/Reactant = (δAXProduct + 1)/(δAXReactant + 1)
Fractionation factors can also be reported using the notation AεProduct/Reactant, which is sometimes called the "enrichment factor" and is calculated as follows:[5]
- AεProduct/Reactant = AαProduct/Reactant − 1
Like δ values, ε values can be reported in per mille by multiplying by 1000.
Δ33S and Δ36S notation
All kinetic and equilibrium isotope effects result from differences in atomic mass.[3][5] As a result, a reaction that fractionates 34S will also fractionate 33S and 36S, and the fractionation factor for each isotope will be mathematically proportional to its mass.[3] Because of the mathematical relationships of their masses, the observed relationships between δ34S, δ33S, and δ36S in most natural materials are approximately δ33S = 0.515 × δ34S and δ36S = 1.90 × δ34S.[8] Rarely, natural processes can create deviations from this relationship, and these deviations are reported as Δ33S and Δ36S values, usually pronounced as "cap delta." These values are typically calculated as follows:[3][9]
- Δ33S = 1000 × [(1 + δ33S/1000) − (1 + δ34S 1000)0.518 − 1]
- Δ36S = 1000 × [(1 + δ36S/1000) − (1 + δ34S/1000)1.91 − 1]
However, the method for calculating Δ33S and Δ36S values is not standardized, and can differ among publications.[10]
.jpg)
Reference materials
Agreed-upon reference materials are required so that reported δ values are comparable among studies. For the sulfur isotope system, δ34S values are reported on the Vienna-Cañon Diablo Troilite (VCDT) scale.[11] The original CDT scale was based on a sample of the mineral troilite recovered from the Canyon Diablo meteorite at Meteor Crater, Arizona, US.[3] The Cañon Diablo Troilite was assigned a δ34S value of 0‰.[3] However, troilite from the Canyon Diablo meteorite was later discovered to have variable sulfur isotope composition.[12] As a result, VCDT was established as a hypothetical sulfur isotope reference with a 34R value of 0.044151[11] and δ34S of 0‰, but no physical sample of VCDT exists. Samples are now measured in comparison to International Atomic Energy Agency (IAEA) reference materials, which are well-characterized, lab-prepared compounds with known δ34S values.[13] A commonly-used IAEA reference material is IAEA-S-1, a silver sulfide reference material with a δ34S value of −0.30‰ VCDT.[4][14] 33S and 36S abundance can also be measured relative to IAEA reference materials and reported on the VCDT scale.[13] For these isotopes, too, VCDT is established as having δ33S and δ36S values of 0‰.[13] The 33R value of VCDT is 0.007877 and the 36R value is 0.0002.[13] IAEA-S-1 has a 33R value of 0.0007878 and a δ33S value of −0.05‰ VCDT; it has a δ36S value of −0.6‰ VCDT.[13]
Analytical methods and instrumentation
The sulfur isotopic composition of natural samples can be determined by Elemental Analysis-Isotope Ratio Mass Spectrometry (EA-IRMS),[15][16] by Dual Inlet-Isotope Ratio Mass Spectrometry (DI-IRMS),[17] by Multi-Collector-Inductively Coupled Plasma Mass Spectrometry (MC-ICPMS),[18] by Secondary Ion Mass Spectrometry (SIMS),[1][19] or by Nanoscale secondary ion mass spectrometry (NanoSIMS).[20] MC-ICPMS can be paired with gas chromatography (GC-MC-ICPMS) to separate certain volatile compounds in a sample and measure the sulfur isotopic composition of individual compounds.[21][22]
The sulfur isotopic compositions of minerals and porewater in sediment are subject to accumulation and diffusion after burial. Reactive transport models are often used to account for the effect of such physical processes and find out the isotopic effect of the process studied.[23][24][25]
Natural variations in sulfur isotope abundance
Sulfur in natural materials

Sulfur is present in the environment in solids, gases, and aqueous species. Sulfur-containing solids on Earth include the common minerals pyrite (FeS2), galena (PbS), and gypsum (CaSO4•2H2O). Sulfur is also an important component of biological material, including in the essential amino acids cysteine and methionine, the B vitamins thiamine and biotin, and the ubiquitous substrate coenzyme A. In the ocean and other natural waters, sulfur is abundant as dissolved sulfate. Hydrogen sulfide is also present in some parts of the deep ocean where it is released from hydrothermal vents. Both sulfate and sulfide can be used by specialized microbes to obtain energy or to grow.[26] Gases including sulfur dioxide and carbonyl sulfide make up the atmospheric component of the sulfur cycle. Any process that transports or chemically transforms sulfur between these many natural materials also has the potential to fractionate sulfur isotopes.
Sulfur isotopic abundance in natural materials

Sulfur in natural materials can vary widely in isotopic composition: compilations of the δ34S values of natural sulfur-containing materials include values ranging from −55‰ to 135‰ VCDT.[27] The ranges of δ34S values vary across sulfur-containing materials: for example, the sulfur in animal tissue ranges from ~ −10 to +20‰ VCDT, while the sulfate in natural waters ranges from ~ −20 to +135‰ VCDT.[27] The range of sulfur isotope abundances in different natural materials results from the isotope fractionation associated with natural processes like the formation and modification of those materials, discussed in the next section.
Processes that fractionate sulfur isotopes
Numerous natural processes are capable of fractionating sulfur isotopes. Microbes are capable of a wide variety of sulfur metabolisms, including the oxidation, reduction, and disproportionation (or simultaneous oxidation and reduction) of sulfur compounds.[1] The effect of these metabolisms on sulfur isotopic composition of the reactants and products is also highly variable, depending on the rate of relevant reactions, availability of nutrients, diagenesis, and other biological, physical and environmental parameters.[25][28][29] As an example, the microbial reduction of sulfate to sulfide generally results in a 34S-depleted product, but the strength of this fractionation has been shown to range from 0 to 65.6‰ VCDT.[28][30]
Many abiotic processes also fractionate sulfur isotopes. Small fractionations with ε values from 0–5‰ have been observed in the formation of the mineral gypsum, an evaporite mineral produced through the evaporation of seawater.[31] Some sulfide minerals, including pyrite and galena, can form through thermochemical sulfate reduction, a process in which seawater sulfate trapped in seafloor rock is reduced to sulfide by geological heat as the rock is buried; this process generally fractionates sulfur more strongly than gypsum formation.[32]
Prior to the rise of oxygen in Earth's atmosphere (referred to as the Great Oxidation Event), additional sulfur-fractionating processes referred to as mass-anomalous or mass-independent fractionation uniquely affected the abundance of 33S and 36S in the rock record.[9] Mass-anomalous fractionations are rare, but they can occur through certain photochemical reactions of gases in the atmosphere.[33][34] Studies have shown that photochemical reactions of atmospheric sulfur dioxide can cause substantial mass-anomalous fractionation of sulfur isotopes.[33][34]
| Process | Range of observed 34ε (‰ VCDT) | Reference |
|---|---|---|
| Assimilatory sulfate reduction | −2.8 to −0.9 | [35][36][37][38] |
| Dissimilatory sulfate reduction | −65.6 to 0 | [28][30][39][40][41][42][43][44][45][46][47][48] |
| Sulfite reduction | −41 to +0.3 | [35][41][49] |
| Sulfide oxidation | −18.0 to +3 | [35][50][51][52][53][54] |
| Sulfur disproportionation | Sulfate: −0.6 to +20.2
Sulfide: −8.6 to −5.5 |
[55][56][57] |
| Thermochemical sulfate reduction | +10 to +25 | [32][58][59] |
| Gypsum formation | 0 to +4.2 | [31][60][61] |
Biological processes of sulfur uptake
All organisms metabolize sulfur, and it is incorporated into the structure of proteins, polysaccharides, steroids, and many coenzymes.[62] The biological pathway by which an organism takes up and/or removes sulfur can have significant impacts on the sulfur isotope composition of the organism and its environment.

Microorganisms that consume and reduce sulfate in relatively large quantities perform a different pathway of sulfur uptake called dissimilatory sulfate reduction. These organisms use sulfate reduction as an energy source as opposed to a way to synthesize new cell components, and remove the resulting sulfide as a waste product. Microbial sulfate reduction has been demonstrated to fractionate sulfur isotopes in bacteria, with some studies showing a dependence upon sulfate concentration[28] and/or temperature.[64] Studies examining dozens of species of dissimilatory sulfate reducing microbes have observed sulfur isotope fractionations ranging from −65.6‰ to 0‰.[28][30][39][40][41][42][43][44][45][46][47][48]

Some organisms take in relatively small amounts of sulfate in a process called assimilatory sulfate reduction, for the purpose of synthesizing compounds that contain sulfur, such as the amino acids methionine and cysteine that can then be used to make proteins.[65] In phytoplankton, most of the sulfur taken up through assimilatory sulfate reduction is incorporated into biomass as proteins (~35%), sulfate esters (~20%), and low-weight sulfur-containing compounds (~40%).[66][67] Literature on the isotopic fractionation effects of the assimilatory sulfate reduction pathway is much more limited than that discussing dissimilatory sulfate reduction, but some sources report slight isotopic variations (δ34S = −4.4‰ to +0.5‰) in the resulting organic sulfur relative to the surrounding sulfate.[68]
While dissimilatory sulfate reduction and assimilatory sulfate reduction are two of the most common pathways by which organisms take up and utilize sulfate, there are many other pathways by which living things take up sulfur. For example, sulfur oxidation of compounds like hydrogen sulfide and elemental sulfur is performed by lithotrophic bacteria and chemosynthetic archaea.[69][70] Most animals obtain sulfur directly from the methionine and cysteine in the protein they consume.[71]
Sulfur isotopes in plants
Methods of detection
Previous efforts to understand how sulfur metabolism and biosynthetic pathways relied on expensive labeling experiments using radioactive 35S. By leveraging natural assimilatory processes, stable isotope ratios can be used to track the sources of sulfur for plants, plant organs used in sulfur acquisition, the movement of sulfur through plants.

Sulfur (S) stable isotope composition measurements are often performed using an Elemental Analysis-Isotope Ratio Mass Spectrometer, (EA-IRMS)[72] in which organic sulfur from biological samples is oxidized to sulfur dioxide (SO2) and analyzed on a mass spectrometer. The mass spectrometer is used to quantify the ratio of the lighter (32S16O2) to the heavier (34S16O2) isotopologue of SO2, and this ratio is then compared to sulfur isotope standards in order to standardize data to the VSMOW scale. In biological materials, sulfur is scarce relative to other organic elements like carbon and oxygen, introducing some additional difficulty in measuring its stable isotope composition. The elemental S composition of plant matter is ≈0.2%, accounting for approximately 2 mmol/m2 in most leaf tissue.[73] In order to reach detectable levels of 30 ng to 3 µg of elemental S to calculate reliable δ34S values, leaf tissue samples need to be between 2–5 mg.
Improvements in detection have been made in recent years in the utilization of gas chromatography coupled with multicollector ICP-MS (GC/MC-ICP-MS)[74] to be able to measure pmol quantities of organic S. Additionally, ICP-MS has been used to measure nanomolar quantities of dissolved sulfate.[75] Most studies have focused on measuring the bulk δ34S value of plant tissues and few studies have been performed on measuring the δ34S values of individual S-containing compounds. The coupling of high-performance liquid chromatography (HPLC) with ICP-MS has been proposed as a way to test individual S-containing compounds.[73]
Sources of sulfur in plants
Each year, approximately 0.3 gigatons of elemental sulfur is converted into organic matter by photosynthetic organisms.[76] This organic sulfur is allocated into a diversity of compounds such as amino acids – namely cysteine (Cys) and methionine (Met) – proteins, cofactors, antioxidants, sulfate groups, Fe-S centers and secondary metabolites. The three main sources of sulfur are atmospheric, soil, and aquatic.
Most vegetation can acquire sulfur from gaseous atmospheric compounds or various ions either in soil solutions or water bodies.[77] Uptake of gaseous and dissolved sulfur compounds apparently occurs with little accompanying isotopic selectivity.[78] Dissolved sulfate (SO42-) is considered to be the central pool which is metabolized by microorganisms and plants as most forms of atmospheric sulfur is oxidized into sulfate. Atmospheric sulfur is eventually returned to the soil when it is scrubbed from the atmosphere during precipitation or through dryfall.[77]
Atmosphere
Many plants acquire sulfur through gaseous atmospheric compounds. Leaves of trees have δ34S values lying between those of air and soil, suggesting that there is uptake occurring from atmospheric and soil sources. The δ34S values of trees has also been demonstrated to be height dependent, with the foliage at the tops of conifers, bull rushes and deciduous trees having δ34S values more reflective of the atmosphere and lower foliage having δ34S values closer to that of soil.[77] It has been proposed that this is due to upper foliage exerting a canopy action on the lower branches, taking up atmospheric sulfur before it can reach lower levels. This is further supported with the epiphytic lichens and mosses having δ34S values close to atmospheric S compounds. This occurs due to lichens and mosses having no access to soil and relying on the direct uptake of gaseous sulfur, dissolved sulfur through rainfall and dry fall accumulation, providing a cumulative record of atmospheric sulfur isotope composition.[77][79]
Main forms of atmospheric sulfur come from the natural sulfur emissions formed biologically and emitted as H2S or organic sulfur gases such as DMS (dimethyl sulfide), COS (carbonyl sulfide), and CS2 (carbon disulfide). These gases are predominantly formed over oceans, wetlands, salt marshes, and estuaries by algae and bacteria.[80] Anthropogenic emissions have increased the concentration of sulfur in the atmosphere mainly through emissions of SO2, from coal, oil, industrial processes, and biomass burning. In 2000, global anthropogenic emission of sulfur was estimated of 55.2–68 Tg S per year, which is much higher than the natural sulfur emissions estimated to be 34 Tg S per year.[80] In the event of excess sulfur in plant tissue, it has been demonstrated that when exposed to high doses of sulfur dioxide, plants emit hydrogen sulfide (H2S) and possibly other reduced sulfur compounds in response to high sulfur loading[78]
Soil
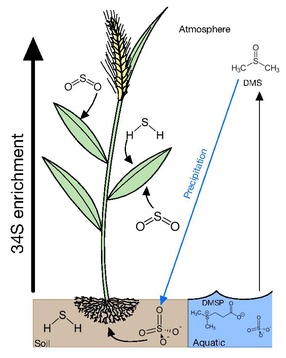
If soil sulfur is derived consistently from one source, the water-soluble and insoluble organic S fractions acquire similar isotopic compositions. In the case that there are two or more sources and/or if the isotopic composition of atmospheric or groundwater sulfate fluctuates, there may not be sufficient time for isotopic homogenization among the various forms of sulfur. The primary form of sulfur in soil is sulfate, which is transported upwards through the root system with minimal δ34S fractionation by 1–2‰.[81] In contrast to higher canopy plants reflecting atmospheric δ34S, protected understory plants tend to reflect soil sulfur.[77]
Aquatic
The forms of sulfur available in aquatic environments depends on whether it is a marine or freshwater environment. Freshwater environments are more varied and subject to a multitude of sulfur inputs and outputs, including atmospheric deposition, runoff, diagenesis of bedrock and the presence of microbial sulfate reducers (MSR). Overall, the main species of sulfur in freshwater environments are hydrogen sulfide and sulfate. In estuaries, plant roots extend into sulfide-rich, 34S-depleted sediments created by MSR, and incorporate that sulfide into their biomass. However, levels of sulfide produced by MSR can be toxic, and it has been proposed that these plants pump oxygen into their roots to oxidize sulfide into the less toxic sulfate.[78] In these environments algae will preferentially acquire sulfur from HS− if present, rather than the more abundant sulfate, as sulfide can be readily incorporated into the direct formation of cysteine. This is consistent with cyanobacteria being able to carry out anoxygenic photosynthesis using sulfide.[77]
In marine environments, the main forms of sulfur available is in sulfate at ~29 mM and a δ34S of 21‰ in seawater. At the surface of the sea, this excess in sulfur is subsequently converted into dimethylsulfoniopropionate (DMSP) by algae as an osmolyte and a repellent against grazing. DMSP also accounts for 50–100% of bacterial sulfur demand, making it the most important source of reduced sulfur for marine bacteria. DMSP's cleavage product dimethyl sulfide (DMS) is highly volatile, escaping the ocean into the atmosphere with emissions ranging between 15 and 33 Tg S year−1 [80] and accounting for 50–60% of the total natural reduced sulfur flux to the atmosphere. In seafloor sediments, microbial sulfate reduction is a major biogeochemical process that consumes organic carbon.[82] Microbial sulfate reduction can completely use up sulfate from the seawater and accumulate hydrogen sulfide in the sediment. Sulfide reoxidation and disproportionation are also thought to be major processes affecting the sulfur isotopic compositions of marine minerals and sediment porewater.[1][25]
Biochemistry
~90% of the organic sulfur in plants is concentrated in the amino acids cysteine and methionine.[81] Cysteine acts as the direct or indirect precursor to any other organic sulfur compounds in plants such as coenzyme-A, methionine, biotin, lipoic acid and glutathione.[78] The carbon skeleton necessary for sulfur assimilation are provided by glycolysis (acetyl-CoA), respiration (aspartic acid, Asp, which derives from oxaloacetate) and photorespiration (serine, Ser).[73] Because cysteine is a direct precursor to methionine, methionine is naturally 34S-depleted in comparison to cysteine.[73] The majority of sulfur is generally in the organic form but, when excess sulfur is available in the environment, inorganic sulfate becomes the major sulfur form. In most plants, 34S discrimination is minimal, and in a study of rice plants it was observed that discrimination takes place in the uptake stage, depleting imported sulfate by 1–2‰ from the source.[83] This effect is through the expression of SO42− transporter genes (SULTR), 14 of which have been identified – which are expressed dependent on the availability of sulfate in the environment. When sulfate is plentiful low affinity transporters are expressed and when sulfate is scarce high affinity genes with greater 34S discrimination are expressed.[73][83]
Distribution through plant organs
Sulfate transported through the roots and SO2 diffusing into leaves becomes the pool for plants to assimilate sulfur throughout their tissues. Though there is minimal fractionation from the source sulfur of the total plant organic matter, in wheat, roots and stems are depleted from soil by 2‰ and leaves and grain are 2‰ enriched. The 34S enrichment in leaf whole matter is not caused by 34S-enriched sulfate present in the leaf, but is the result of the 34S-enrichment arriving at sink organs causing proteins in the leaves to be 34S-enriched.[73] In rice, translocation from root to shoot does not discriminate S isotopes, however, the sulfate pools of the shoot are significantly 34S-enriched with respect to the sulfate pools of both root and sap. As sulfate moves through the plant system and is incorporated into biomass, the pool becomes enriched, giving organs such as leaves and grains higher δ34S values than earlier tissues.[83]
Applications
Rise of atmospheric oxygen
Signatures of mass-anomalous sulfur isotope fractionation preserved in the rock record have been an important piece of evidence for understanding the Great Oxidation Event, the sudden rise of oxygen on the ancient Earth.[9][84] Nonzero values of Δ33S and Δ36S are present in the sulfur-bearing minerals of Precambrian rock formed greater than 2.45 billion years ago, but completely absent from rock less than 2.09 billion years old.[9] Multiple mechanisms have been proposed for how oxygen prevents the fingerprints of mass-anomalous fractionation from being created and preserved; nevertheless, all studies of Δ33S and Δ36S records conclude that oxygen was essentially absent from Earth's atmosphere prior to 2.45 billion years ago.[9][10][33][85][86]
Paleobiology and paleoclimate
A number of microbial metabolisms fractionate sulfur isotopes in distinctive ways, and the sulfur isotopic fingerprints of these metabolisms can be preserved in minerals and ancient organic matter.[1] By measuring the sulfur isotopic composition of these preserved materials, scientists can reconstruct ancient biological processes and the environments where they occurred.[1] δ34S values in the geologic record have been inferred to reveal the history of microbial sulfate reduction[87][88] and sulfide oxidation.[25][89] Paired δ34S and Δ33S records have also been used to show ancient microbial sulfur disproportionation.[90][29]

Microbial dissimilatory sulfate reduction (MSR), an energy-yielding metabolism performed by bacteria in anoxic environments, is associated with an especially large fractionation factor.[1] The observed 34εMSR values range from 0 to −65.6‰.[28][30][39][40][41][42][43][44] Many factors influence the size of this fractionation, including sulfate reduction rate,[35][40] sulfate concentration and transport,[28][44] availability of electron donors and other nutrients,[30][42][43] and physiological differences like protein expression.[45] Sulfide produced through MSR may then go on to form the mineral pyrite, preserving the 34S-depleted fingerprint of MSR in sedimentary rocks.[1][60] Many studies have investigated the δ34S values of ancient pyrite in order to understand past biological and environmental conditions.[1] For example, pyrite δ34S records have been used to reconstruct shifts in primary productivity levels,[91] changing ocean oxygen content,[92][93] and glacial-interglacial changes in sea level and weathering.[94] Some studies compare sulfur isotopes in pyrite to a second sulfur-containing material, like dissolved sulfate or preserved organic matter.[25][91][92] Comparing pyrite to another material gives a fuller picture of how sulfur moved through ancient environments: it provides clues about the size of ancient 34εMSR values and the environmental conditions controlling MSR fractionation of sulfur isotopes.[91][92]
Paleoceanography
δ34S records have been used to infer changes in seawater sulfate concentrations.[95] Because the δ34S values of carbonate-associated sulfate are thought to be sensitive to seawater sulfate levels, these measurements have been used to reconstruct the history of seawater sulfate.[96] δ34S values of pyrite have also been applied to reconstruct the concentration of seawater sulfate, based on expected biological fractionations at low sulfate concentrations.[97][98] Both of these methods rely on assumptions about the depositional environment or the biological community, creating some uncertainty in the resulting reconstructions.[28][96]
See also
- Δ34S
- Isotopes of sulfur
- Hydrogen isotope biogeochemistry
- Stable isotope ratio
- Isotope analysis
- Isotope geochemistry
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Canfield, D. E. (2001-01-01). "Biogeochemistry of Sulfur Isotopes" (in en). Reviews in Mineralogy and Geochemistry 43 (1): 607–636. doi:10.2138/gsrmg.43.1.607. ISSN 1529-6466. Bibcode: 2001RvMG...43..607C. https://pubs.geoscienceworld.org/rimg/article-abstract/43/1/607/140690/Biogeochemistry-of-Sulfur-Isotopes.
- ↑ 2.0 2.1 Wang, Meng; Audi, G.; Kondev, F. G.; Huang, W.J.; Naimi, S.; Xu, Xing (2017). "The AME2016 atomic mass evaluation (II). Tables, graphs and references" (in en). Chinese Physics C 41 (3): 030003. doi:10.1088/1674-1137/41/3/030003. ISSN 1674-1137. Bibcode: 2017ChPhC..41c0003W.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 Sharp, Zachary (2006). Principles of Stable Isotope Geochemistry.
- ↑ 4.0 4.1 Brand, Willi; Coplen, Tyler; Vogel, Jochen; Rosner, Martin; Prohaska, Thomas (2014). "Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)". Pure and Applied Chemistry 86 (3): 425–467. doi:10.1515/pac-2013-1023. https://www.degruyter.com/view/journals/pac/86/3/article-p425.xml.
- ↑ 5.00 5.01 5.02 5.03 5.04 5.05 5.06 5.07 5.08 5.09 5.10 5.11 5.12 5.13 5.14 5.15 Hayes, John (2002). Practices and Principles of Stable Isotopic Measurement in Organic Geochemistry (Revision 2). https://www.researchgate.net/publication/255578228.
- ↑ Mccready, R. G. L.; Laishley, E. J.; Krouse, H. R. (1976-08-01). "Biogeochemical implications of inverse sulfur isotope effects during reduction of sulfur compounds by Clostridium pasteurianum" (in en). Geochimica et Cosmochimica Acta 40 (8): 979–981. doi:10.1016/0016-7037(76)90146-0. ISSN 0016-7037. Bibcode: 1976GeCoA..40..979M.
- ↑ Craig, H. and Gordon, L. I. 1965. “Deuterium and oxygen 18 variations in the ocean and the marine atmosphere”. In Stable Isotopes in Oceanographic Studies and Paleotemperatures, Edited by: Tongiorgi, E. 9–130. Pisa: Laboratorio di Geologia Nucleare.
- ↑ Hulston, J. R.; Thode, H. G. (1965). "Variations in the S33, S34, and S36 contents of meteorites and their relation to chemical and nuclear effects" (in en). Journal of Geophysical Research 70 (14): 3475–3484. doi:10.1029/JZ070i014p03475. ISSN 2156-2202. Bibcode: 1965JGR....70.3475H.
- ↑ 9.0 9.1 9.2 9.3 9.4 Farquhar, James; Bao, Huiming; Thiemens, Mark (2000-08-04). "Atmospheric Influence of Earth's Earliest Sulfur Cycle" (in en). Science 289 (5480): 756–758. doi:10.1126/science.289.5480.756. ISSN 0036-8075. PMID 10926533. Bibcode: 2000Sci...289..756F. https://www.science.org/doi/10.1126/science.289.5480.756.
- ↑ 10.0 10.1 Halevy, Itay; Johnston, David T.; Schrag, Daniel P. (2010-07-09). "Explaining the Structure of the Archean Mass-Independent Sulfur Isotope Record" (in en). Science 329 (5988): 204–207. doi:10.1126/science.1190298. ISSN 0036-8075. PMID 20508089. Bibcode: 2010Sci...329..204H. https://www.science.org/doi/10.1126/science.1190298.
- ↑ 11.0 11.1 Ding, Tiping; Bai, Ruimei; Li, Yanhe; Wan, Defang; Zou, Xiaoqiu; Zhang, Qinglian (1999-08-01). "Determination of the absolute32S/34S ratio of IAEA-S-1 reference material and V-CDT sulfur isotope standard" (in en). Science in China Series D: Earth Sciences 42 (1): 45–51. doi:10.1007/BF02878497. ISSN 1862-2801. Bibcode: 1999ScChD..42...45D.
- ↑ Beaudoin, Georges; Taylor, B. E.; Rumble, D.; Thiemens, M. (1994-10-01). "Variations in the sulfur isotope composition of troilite from the Cañon Diablo iron meteorite" (in en). Geochimica et Cosmochimica Acta 58 (19): 4253–4255. doi:10.1016/0016-7037(94)90277-1. ISSN 0016-7037. Bibcode: 1994GeCoA..58.4253B.
- ↑ 13.0 13.1 13.2 13.3 13.4 Ding, T.; Valkiers, S.; Kipphardt, H.; De Bièvre, P.; Taylor, P. D. P.; Gonfiantini, R.; Krouse, R. (2001-08-01). "Calibrated sulfur isotope abundance ratios of three IAEA sulfur isotope reference materials and V-CDT with a reassessment of the atomic weight of sulfur" (in en). Geochimica et Cosmochimica Acta 65 (15): 2433–2437. doi:10.1016/S0016-7037(01)00611-1. ISSN 0016-7037. Bibcode: 2001GeCoA..65.2433D.
- ↑ Krouse, H. R.; Coplen, Tyler B. (1997-02-28). "Reporting of relative sulfur isotope-ratio data (Technical Report)" (in en). Pure and Applied Chemistry 69 (2): 293–296. doi:10.1351/pac199769020293. ISSN 0033-4545.
- ↑ Giesemann, A.; Jaeger, H.-J.; Norman, A. L.; Krouse, H. R.; Brand, W. A. (1994-09-15). "Online Sulfur-Isotope Determination Using an Elemental Analyzer Coupled to a Mass Spectrometer". Analytical Chemistry 66 (18): 2816–2819. doi:10.1021/ac00090a005. ISSN 0003-2700.
- ↑ Grassineau, Nathalie V. (2006-05-01). "High-precision EA-IRMS analysis of S and C isotopes in geological materials" (in en). Applied Geochemistry. Frontiers in Analytical Geochemistry–An IGC 2004 Perspective 21 (5): 756–765. doi:10.1016/j.apgeochem.2006.02.015. ISSN 0883-2927. Bibcode: 2006ApGC...21..756G.
- ↑ Mayer, Bernhard; Krouse, H. Roy (2004-01-01), de Groot, Pier A., ed., "Chapter 26 - Procedures for Sulfur Isotope Abundance Studies" (in en), Handbook of Stable Isotope Analytical Techniques (Elsevier): pp. 538–596, ISBN 978-0-444-51114-0, http://www.sciencedirect.com/science/article/pii/B9780444511140500284, retrieved 2020-05-25
- ↑ Paris, Guillaume; Sessions, Alex L.; Subhas, Adam V.; Adkins, Jess F. (2013-05-08). "MC-ICP-MS measurement of δ34S and ∆33S in small amounts of dissolved sulfate" (in en). Chemical Geology 345: 50–61. doi:10.1016/j.chemgeo.2013.02.022. ISSN 0009-2541. Bibcode: 2013ChGeo.345...50P. http://www.sciencedirect.com/science/article/pii/S0009254113000818.
- ↑ Riciputi, Lee R; Paterson, Bruce A; Ripperdan, Robert L (1998-10-19). "Measurement of light stable isotope ratios by SIMS:: Matrix effects for oxygen, carbon, and sulfur isotopes in minerals33Dedicated to the memory of Al Nier." (in en). International Journal of Mass Spectrometry 178 (1): 81–112. doi:10.1016/S1387-3806(98)14088-5. ISSN 1387-3806.
- ↑ Hauri, Erik H.; Papineau, Dominic; Wang, Jianhua; Hillion, Francois (2016-01-20). "High-precision analysis of multiple sulfur isotopes using NanoSIMS" (in en). Chemical Geology 420: 148–161. doi:10.1016/j.chemgeo.2015.11.013. ISSN 0009-2541. Bibcode: 2016ChGeo.420..148H. https://discovery.ucl.ac.uk/id/eprint/1474747/1/Hauri_et_al_2016_ChemGeol_Multiple_Sisotopes_NS.pdf.
- ↑ Amrani, Alon; Sessions, Alex L.; Adkins, Jess F. (2009-11-01). "Compound-Specific δ34S Analysis of Volatile Organics by Coupled GC/Multicollector-ICPMS". Analytical Chemistry 81 (21): 9027–9034. doi:10.1021/ac9016538. ISSN 0003-2700. PMID 19807109.
- ↑ Said‐Ahmad, Ward; Amrani, Alon (2013). "A sensitive method for the sulfur isotope analysis of dimethyl sulfide and dimethylsulfoniopropionate in seawater" (in en). Rapid Communications in Mass Spectrometry 27 (24): 2789–2796. doi:10.1002/rcm.6751. ISSN 1097-0231. PMID 24214865. Bibcode: 2013RCMS...27.2789S.
- ↑ Berner, Robert A. (31 March 2020). Early Diagenesis : A Theoretical Approach. Princeton University Press. ISBN 978-0-691-20940-1. OCLC 1164642477. http://worldcat.org/oclc/1164642477.
- ↑ J⊘rgensen, Bo Barker (January 1978). "A comparison of methods for the quantification of bacterial sulfate reduction in coastal marine sediments". Geomicrobiology Journal 1 (1): 29–47. doi:10.1080/01490457809377722. ISSN 0149-0451. http://dx.doi.org/10.1080/01490457809377722.
- ↑ 25.0 25.1 25.2 25.3 25.4 Tsang, Man-Yin; Wortmann, Ulrich G. (2022-07-06). "Sulfur isotope fractionation derived from reaction-transport modelling in the Eastern Equatorial Pacific". Journal of the Geological Society 179 (5). doi:10.1144/jgs2021-068. ISSN 0016-7649. https://doi.org/10.1144/jgs2021-068.
- ↑ Canfield, Donald E.; Erik Kristensen; Bo Thamdrup (2005-01-01), Canfield, Donald E.; Kristensen, Erik; Thamdrup, Bo, eds., "The Sulfur Cycle" (in en), Advances in Marine Biology, Aquatic Geomicrobiology (Academic Press) 48: pp. 313–381, doi:10.1016/S0065-2881(05)48009-8, ISBN 9780120261475, http://www.sciencedirect.com/science/article/pii/S0065288105480098, retrieved 2020-05-23
- ↑ 27.0 27.1 Meija, Juris; Coplen, Tyler; Berglund, Michael; Brand, Willi; De Bievre, Paul; Groening, Manfred; Holden, Norman; Irrgeher, Johanna et al. (2013). "Atomic weights of the elements 2013 (IUPAC Technical Report)". Pure and Applied Chemistry 88. https://www.degruyter.com/view/journals/pac/88/3/article-p265.xml.
- ↑ 28.0 28.1 28.2 28.3 28.4 28.5 28.6 28.7 Bradley, A. S.; Leavitt, W. D.; Schmidt, M.; Knoll, A. H.; Girguis, P. R.; Johnston, D. T. (January 2016). "Patterns of sulfur isotope fractionation during microbial sulfate reduction" (in en). Geobiology 14 (1): 91–101. doi:10.1111/gbi.12149. PMID 26189479. http://nrs.harvard.edu/urn-3:HUL.InstRepos:30367425.
- ↑ 29.0 29.1 Johnston, David T.; Farquhar, James; Wing, Boswell A.; Kaufman, Alan J.; Canfield, Donald E.; Habicht, Kirsten S. (2005-06-01). "Multiple sulfur isotope fractionations in biological systems: A case study with sulfate reducers and sulfur disproportionators" (in en). American Journal of Science 305 (6–8): 645–660. doi:10.2475/ajs.305.6-8.645. ISSN 0002-9599. Bibcode: 2005AmJS..305..645J. http://www.ajsonline.org/content/305/6-8/645.
- ↑ 30.0 30.1 30.2 30.3 30.4 Sim, Min Sub; Bosak, Tanja; Ono, Shuhei (2011-07-01). "Large Sulfur Isotope Fractionation Does Not Require Disproportionation" (in en). Science 333 (6038): 74–77. doi:10.1126/science.1205103. ISSN 0036-8075. PMID 21719675. Bibcode: 2011Sci...333...74S. https://www.science.org/doi/10.1126/science.1205103.
- ↑ 31.0 31.1 Raab, M.; Spiro, B. (1991-04-05). "Sulfur isotopic variations during seawater evaporation with fractional crystallization" (in en). Chemical Geology: Isotope Geoscience Section 86 (4): 323–333. doi:10.1016/0168-9622(91)90014-N. ISSN 0168-9622.
- ↑ 32.0 32.1 Machel, Hans G.; Krouse, H. Roy; Sassen, Roger (1995-07-01). "Products and distinguishing criteria of bacterial and thermochemical sulfate reduction" (in en). Applied Geochemistry 10 (4): 373–389. doi:10.1016/0883-2927(95)00008-8. ISSN 0883-2927. Bibcode: 1995ApGC...10..373M.
- ↑ 33.0 33.1 33.2 Masterson, Andrew L.; Farquhar, James; Wing, Boswell A. (2011-06-15). "Sulfur mass-independent fractionation patterns in the broadband UV photolysis of sulfur dioxide: Pressure and third body effects" (in en). Earth and Planetary Science Letters 306 (3): 253–260. doi:10.1016/j.epsl.2011.04.004. ISSN 0012-821X. Bibcode: 2011E&PSL.306..253M. http://www.sciencedirect.com/science/article/pii/S0012821X1100210X.
- ↑ 34.0 34.1 Lyons, James R. (2007). "Mass-independent fractionation of sulfur isotopes by isotope-selective photodissociation of SO2" (in en). Geophysical Research Letters 34 (22): L22811. doi:10.1029/2007GL031031. ISSN 1944-8007. Bibcode: 2007GeoRL..3422811L.
- ↑ 35.0 35.1 35.2 35.3 Kaplan, I. R.; Rittenberg, S. C. (1964). "Microbiological Fractionation of Sulphur Isotopes". Microbiology 34 (2): 195–212. doi:10.1099/00221287-34-2-195. ISSN 1350-0872. PMID 14135528.
- ↑ Trust, B. A.; Fry, B. (1992). "Stable sulphur isotopes in plants: a review" (in en). Plant, Cell & Environment 15 (9): 1105–1110. doi:10.1111/j.1365-3040.1992.tb01661.x. ISSN 1365-3040.
- ↑ Patron, Nicola J.; Durnford, Dion G.; Kopriva, Stanislav (2008-02-04). "Sulfate assimilation in eukaryotes: fusions, relocations and lateral transfers" (in en). BMC Evolutionary Biology 8 (1): 39. doi:10.1186/1471-2148-8-39. ISSN 1471-2148. PMID 18248682.
- ↑ Schiff, J. A.; Fankhauser, H. (1981). Bothe, Hermann; Trebst, Achim. eds. "Assimilatory Sulfate Reduction" (in en). Biology of Inorganic Nitrogen and Sulfur. Proceedings in Life Sciences (Berlin, Heidelberg: Springer): 153–168. doi:10.1007/978-3-642-67919-3_11. ISBN 978-3-642-67919-3.
- ↑ 39.0 39.1 39.2 Detmers, Jan; Brüchert, Volker; Habicht, Kirsten S.; Kuever, Jan (2001-02-01). "Diversity of Sulfur Isotope Fractionations by Sulfate-Reducing Prokaryotes" (in en). Applied and Environmental Microbiology 67 (2): 888–894. doi:10.1128/AEM.67.2.888-894.2001. ISSN 0099-2240. PMID 11157259. Bibcode: 2001ApEnM..67..888D.
- ↑ 40.0 40.1 40.2 40.3 Canfield, D. E. (2001-04-01). "Isotope fractionation by natural populations of sulfate-reducing bacteria" (in en). Geochimica et Cosmochimica Acta 65 (7): 1117–1124. doi:10.1016/S0016-7037(00)00584-6. ISSN 0016-7037. Bibcode: 2001GeCoA..65.1117C. http://www.sciencedirect.com/science/article/pii/S0016703700005846.
- ↑ 41.0 41.1 41.2 41.3 Kemp, A. L. W.; Thode, H. G. (1968-01-01). "The mechanism of the bacterial reduction of sulphate and of sulphite from isotope fractionation studies" (in en). Geochimica et Cosmochimica Acta 32 (1): 71–91. doi:10.1016/0016-7037(68)90088-4. ISSN 0016-7037. Bibcode: 1968GeCoA..32...71K.
- ↑ 42.0 42.1 42.2 42.3 Sim, Min Sub; Ono, Shuhei; Donovan, Katie; Templer, Stefanie P.; Bosak, Tanja (2011-08-01). "Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibrio sp." (in en). Geochimica et Cosmochimica Acta 75 (15): 4244–4259. doi:10.1016/j.gca.2011.05.021. ISSN 0016-7037. Bibcode: 2011GeCoA..75.4244S. http://www.sciencedirect.com/science/article/pii/S0016703711002894.
- ↑ 43.0 43.1 43.2 43.3 Sim, Min Sub; Ono, Shuhei; Bosak, Tanja (2012-12-01). "Effects of Iron and Nitrogen Limitation on Sulfur Isotope Fractionation during Microbial Sulfate Reduction" (in en). Applied and Environmental Microbiology 78 (23): 8368–8376. doi:10.1128/AEM.01842-12. ISSN 0099-2240. PMID 23001667. Bibcode: 2012ApEnM..78.8368S.
- ↑ 44.0 44.1 44.2 44.3 Sim, Min Sub; Paris, Guillaume; Adkins, Jess F.; Orphan, Victoria J.; Sessions, Alex L. (2017-06-01). "Quantification and isotopic analysis of intracellular sulfur metabolites in the dissimilatory sulfate reduction pathway" (in en). Geochimica et Cosmochimica Acta 206: 57–72. doi:10.1016/j.gca.2017.02.024. ISSN 0016-7037. Bibcode: 2017GeCoA.206...57S. http://www.sciencedirect.com/science/article/pii/S0016703717301187.
- ↑ 45.0 45.1 45.2 Leavitt, William D.; Venceslau, Sofia S.; Pereira, Inês A. C.; Johnston, David T.; Bradley, Alexander S. (2016-10-01). "Fractionation of sulfur and hydrogen isotopes in Desulfovibrio vulgaris with perturbed DsrC expression" (in en). FEMS Microbiology Letters 363 (20): fnw226. doi:10.1093/femsle/fnw226. ISSN 0378-1097. PMID 27702753. https://academic.oup.com/femsle/article/363/20/fnw226/2680364.
- ↑ 46.0 46.1 Detmers, Jan; Brüchert, Volker; Habicht, Kirsten S.; Kuever, Jan (February 2001). "Diversity of Sulfur Isotope Fractionations by Sulfate-Reducing Prokaryotes" (in en). Applied and Environmental Microbiology 67 (2): 888–894. doi:10.1128/AEM.67.2.888-894.2001. ISSN 0099-2240. PMID 11157259. Bibcode: 2001ApEnM..67..888D.
- ↑ 47.0 47.1 Rees, C. E. (1973-05-01). "A steady-state model for sulphur isotope fractionation in bacterial reduction processes" (in en). Geochimica et Cosmochimica Acta 37 (5): 1141–1162. doi:10.1016/0016-7037(73)90052-5. ISSN 0016-7037. Bibcode: 1973GeCoA..37.1141R. https://dx.doi.org/10.1016/0016-7037%2873%2990052-5.
- ↑ 48.0 48.1 Habicht, Kirsten S.; Canfield, Donald E. (1997-12-01). "Sulfur isotope fractionation during bacterial sulfate reduction in organic-rich sediments" (in en). Geochimica et Cosmochimica Acta 61 (24): 5351–5361. doi:10.1016/S0016-7037(97)00311-6. ISSN 0016-7037. PMID 11541664. Bibcode: 1997GeCoA..61.5351H. https://www.sciencedirect.com/science/article/pii/S0016703797003116.
- ↑ Leavitt, William D.; Cummins, Renata; Schmidt, Marian L.; Sim, Min S.; Ono, Shuhei; Bradley, Alexander S.; Johnston, David T. (2014). "Multiple sulfur isotope signatures of sulfite and thiosulfate reduction by the model dissimilatory sulfate-reducer, Desulfovibrio alaskensis str. G20" (in en). Frontiers in Microbiology 5: 591. doi:10.3389/fmicb.2014.00591. ISSN 1664-302X. PMID 25505449.
- ↑ Balci, Nurgul; Shanks, Wayne C.; Mayer, Bernhard; Mandernack, Kevin W. (2007-08-01). "Oxygen and sulfur isotope systematics of sulfate produced by bacterial and abiotic oxidation of pyrite" (in en). Geochimica et Cosmochimica Acta 71 (15): 3796–3811. doi:10.1016/j.gca.2007.04.017. ISSN 0016-7037. Bibcode: 2007GeCoA..71.3796B. http://www.sciencedirect.com/science/article/pii/S0016703707002141.
- ↑ Howard Gest, Brian Fry; Hayes, J. M. (1984-05-01). "Isotope effects associated with the anaerobic oxidation of sulfide by the purple photosynthetic bacterium, Chromatium vinosum" (in en). FEMS Microbiology Letters 22 (3): 283–287. doi:10.1111/j.1574-6968.1984.tb00742.x. ISSN 0378-1097.
- ↑ Jones, Galen E.; Starkey, Robert L. (March 1957). "Fractionation of Stable Isotopes of Sulfur by Microorganisms and Their Role in Deposition of Native Sulfur". Applied Microbiology 5 (2): 111–118. doi:10.1128/AEM.5.2.111-118.1957. ISSN 0003-6919. PMID 13425528.
- ↑ Kaplan, I. R.; Rafter, T. A. (1958-03-07). "Fractionation of Stable Isotopes of Sulfur by Thiobacilli" (in en). Science 127 (3297): 517–518. doi:10.1126/science.127.3297.517. ISSN 0036-8075. PMID 13529013. Bibcode: 1958Sci...127..517K. https://www.science.org/doi/10.1126/science.127.3297.517.
- ↑ Nakai, Nobuyuki; Jensen, M. L. (1964-12-01). "The kinetic isotope effect in the bacterial reduction and oxidation of sulfur" (in en). Geochimica et Cosmochimica Acta 28 (12): 1893–1912. doi:10.1016/0016-7037(64)90136-X. ISSN 0016-7037. Bibcode: 1964GeCoA..28.1893N.
- ↑ Canfield, D. E.; Thamdrup, B. (1994-12-23). "The production of 34S-depleted sulfide during bacterial disproportionation of elemental sulfur" (in en). Science 266 (5193): 1973–1975. doi:10.1126/science.11540246. ISSN 0036-8075. PMID 11540246. Bibcode: 1994Sci...266.1973C. https://www.science.org/doi/10.1126/science.11540246.
- ↑ Canfield, D. E.; Thamdrup, B.; Fleischer, S. (1998). "Isotope fractionation and sulfur metabolism by pure and enrichment cultures of elemental sulfur-disproportionating bacteria" (in en). Limnology and Oceanography 43 (2): 253–264. doi:10.4319/lo.1998.43.2.0253. ISSN 1939-5590. Bibcode: 1998LimOc..43..253C.
- ↑ Böttcher, Michael E; Thamdrup, Bo (2001-05-15). "Anaerobic sulfide oxidation and stable isotope fractionation associated with bacterial sulfur disproportionation in the presence of MnO2" (in en). Geochimica et Cosmochimica Acta 65 (10): 1573–1581. doi:10.1016/S0016-7037(00)00622-0. ISSN 0016-7037. Bibcode: 2001GeCoA..65.1573B. http://www.sciencedirect.com/science/article/pii/S0016703700006220.
- ↑ Amrani, Alon; Deev, Andrei; Sessions, Alex L.; Tang, Yongchun; Adkins, Jess F.; Hill, Ronald J.; Moldowan, J. Michael; Wei, Zhibin (2012-05-01). "The sulfur-isotopic compositions of benzothiophenes and dibenzothiophenes as a proxy for thermochemical sulfate reduction" (in en). Geochimica et Cosmochimica Acta 84: 152–164. doi:10.1016/j.gca.2012.01.023. ISSN 0016-7037. Bibcode: 2012GeCoA..84..152A. http://www.sciencedirect.com/science/article/pii/S0016703712000464.
- ↑ Kiyosu, Yasuhiro; Krouse, H. Roy (1993). "Thermochemical reduction and sulfur isotopic behavior of sulfate by acetic acid in the presence of native sulfur". Geochemical Journal 27 (1): 49–57. doi:10.2343/geochemj.27.49. Bibcode: 1993GeocJ..27...49K. https://www.jstage.jst.go.jp/article/geochemj1966/27/1/27_1_49/_article/-char/ja/.
- ↑ 60.0 60.1 Ault, W. U; Kulp, J. L (1959-07-01). "Isotopic geochemistry of sulphur" (in en). Geochimica et Cosmochimica Acta 16 (4): 201–235. doi:10.1016/0016-7037(59)90112-7. ISSN 0016-7037. Bibcode: 1959GeCoA..16..201A.
- ↑ Thode, H. G; Monster, J; Dunford, H. B (1961-11-01). "Sulphur isotope geochemistry" (in en). Geochimica et Cosmochimica Acta 25 (3): 159–174. doi:10.1016/0016-7037(61)90074-6. ISSN 0016-7037. Bibcode: 1961GeCoA..25..159T.
- ↑ Elliott, Katherine; Whelan, Julie (2009-09-16) (in en). Sulphur in Biology. John Wiley & Sons. ISBN 978-0-470-71823-0. https://books.google.com/books?id=YxosoxZi-t0C&dq=Pathways+of+assimilatory+sulphate+reduction+in+plants+and+microorganisms.&pg=PA49.
- ↑ 63.0 63.1 Santos, André A.; Venceslau, Sofia S.; Grein, Fabian; Leavitt, William D.; Dahl, Christiane; Johnston, David T.; Pereira, Inês A. C. (2015-12-18). "A protein trisulfide couples dissimilatory sulfate reduction to energy conservation" (in en). Science 350 (6267): 1541–1545. doi:10.1126/science.aad3558. ISSN 0036-8075. PMID 26680199. Bibcode: 2015Sci...350.1541S. https://www.science.org/doi/10.1126/science.aad3558.
- ↑ Canfield, Donald E.; Olesen, Claus A.; Cox, Raymond P. (2006-02-01). "Temperature and its control of isotope fractionation by a sulfate-reducing bacterium" (in en). Geochimica et Cosmochimica Acta 70 (3): 548–561. doi:10.1016/j.gca.2005.10.028. ISSN 0016-7037. Bibcode: 2006GeCoA..70..548C. https://www.sciencedirect.com/science/article/pii/S0016703705008100.
- ↑ "5.9C: Sulfate and Sulfur Reduction" (in en). 2017-05-09. https://bio.libretexts.org/Bookshelves/Microbiology/Book%3A_Microbiology_(Boundless)/5%3A_Microbial_Metabolism/5.09%3A_Anaerobic_Respiration/5.9C%3A_Sulfate_and_Sulfur_Reduction.
- ↑ Bates, Timothy S.; Kiene, Ronald P.; Wolfe, Gordon V.; Matrai, Patricia A.; Chavez, Francisco P.; Buck, Kurt R.; Blomquist, Byron W.; Cuhel, Russell L. (1994). "The cycling of sulfur in surface seawater of the northeast Pacific" (in en). Journal of Geophysical Research 99 (C4): 7835. doi:10.1029/93JC02782. ISSN 0148-0227. Bibcode: 1994JGR....99.7835B. http://doi.wiley.com/10.1029/93JC02782.
- ↑ Giordano, Mario; Norici, Alessandra; Hell, Ruediger (May 2005). "Sulfur and phytoplankton: acquisition, metabolism and impact on the environment" (in en). New Phytologist 166 (2): 371–382. doi:10.1111/j.1469-8137.2005.01335.x. ISSN 0028-646X. PMID 15819903. https://onlinelibrary.wiley.com/doi/10.1111/j.1469-8137.2005.01335.x.
- ↑ Chambers, L. A.; Trudinger, P. A. (January 1979). "Microbiological fractionation of stable sulfur isotopes: A review and critique" (in en). Geomicrobiology Journal 1 (3): 249–293. doi:10.1080/01490457909377735. ISSN 0149-0451. http://www.tandfonline.com/doi/abs/10.1080/01490457909377735.
- ↑ Ghosh, Wriddhiman; Dam, Bomba (2009-11-01). "Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea". FEMS Microbiology Reviews 33 (6): 999–1043. doi:10.1111/j.1574-6976.2009.00187.x. ISSN 0168-6445. PMID 19645821.
- ↑ Paredes, Gabriela F.; Viehboeck, Tobias; Lee, Raymond; Palatinszky, Marton; Mausz, Michaela A.; Reipert, Siegfried; Schintlmeister, Arno; Maier, Andreas et al. (2021-06-29). Bordenstein, Seth. ed. "Anaerobic Sulfur Oxidation Underlies Adaptation of a Chemosynthetic Symbiont to Oxic-Anoxic Interfaces" (in en). mSystems 6 (3): e01186–20. doi:10.1128/mSystems.01186-20. ISSN 2379-5077. PMID 34058098.
- ↑ Komarnisky, Lioudmila A; Christopherson, Robert J; Basu, Tapan K (2003-01-01). "Sulfur: its clinical and toxicologic aspects" (in en). Nutrition 19 (1): 54–61. doi:10.1016/S0899-9007(02)00833-X. ISSN 0899-9007. PMID 12507640. https://www.sciencedirect.com/science/article/pii/S089990070200833X.
- ↑ Grassineau, NV (1998). "Measurement of sulphur isotopic compositions of sulphide minerals using new continuous He-flow EA-MS technology". Mineralogical Magazine 62 (1): 537–538. doi:10.1180/minmag.1998.62A.1.284. Bibcode: 1998MinM...62..537G. https://rruff.info/doclib/MinMag/Volume_62A/62A-1-537.pdf.
- ↑ 73.0 73.1 73.2 73.3 73.4 73.5 Tcherkez, Guillaume; Tea, Illa (October 2013). "32 S/ 34 S isotope fractionation in plant sulphur metabolism" (in en). New Phytologist 200 (1): 44–53. doi:10.1111/nph.12314. ISSN 0028-646X.
- ↑ Amrani, Alon; Sessions, Alex L.; Adkins, Jess F. (2009-11-01). "Compound-Specific δ 34 S Analysis of Volatile Organics by Coupled GC/Multicollector-ICPMS" (in en). Analytical Chemistry 81 (21): 9027–9034. doi:10.1021/ac9016538. ISSN 0003-2700. PMID 19807109. https://pubs.acs.org/doi/10.1021/ac9016538.
- ↑ Paris, Guillaume; Sessions, Alex L.; Subhas, Adam V.; Adkins, Jess F. (2013-05-08). "MC-ICP-MS measurement of δ34S and ∆33S in small amounts of dissolved sulfate" (in en). Chemical Geology 345: 50–61. doi:10.1016/j.chemgeo.2013.02.022. ISSN 0009-2541. Bibcode: 2013ChGeo.345...50P. https://www.sciencedirect.com/science/article/pii/S0009254113000818.
- ↑ Ivanov, M.V. (1981). Some Perspectives of the Major Biogeochemical Cycles. USSR: SCOPE. pp. 61–78.
- ↑ 77.0 77.1 77.2 77.3 77.4 77.5 Krouse, H. R. (1989). "Sulfur Isotope Studies of the Pedosphere and Biosphere". in Rundel, P. W.; Ehleringer, J. R.; Nagy, K. A. (in en). Stable Isotopes in Ecological Research. Ecological Studies. 68. New York, NY: Springer. pp. 424–444. doi:10.1007/978-1-4612-3498-2_24. ISBN 978-1-4612-3498-2. https://link.springer.com/chapter/10.1007/978-1-4612-3498-2_24.
- ↑ 78.0 78.1 78.2 78.3 Trust, B. A.; Fry, B. (December 1992). "Stable sulphur isotopes in plants: a review" (in en). Plant, Cell and Environment 15 (9): 1105–1110. doi:10.1111/j.1365-3040.1992.tb01661.x. ISSN 0140-7791. https://onlinelibrary.wiley.com/doi/10.1111/j.1365-3040.1992.tb01661.x.
- ↑ Wadleigh, Moire A. (2003). "Lichens and atmospheric sulphur: what stable isotopes reveal". Environmental Pollution 126 (3): 345–351. doi:10.1016/s0269-7491(03)00247-1. ISSN 0269-7491. PMID 12963295. https://pubmed.ncbi.nlm.nih.gov/12963295/.
- ↑ 80.0 80.1 80.2 De Kok, Luit J.; Durenkamp, Mark; Yang, Liping; Stulen, Ineke (2007), Hawkesford, Malcolm J.; De Kok, Luit J., eds., "Atmospheric sulfur" (in en), Sulfur in Plants An Ecological Perspective, Plant Ecophysiology (Dordrecht: Springer Netherlands) 6: pp. 91–106, doi:10.1007/978-1-4020-5887-5_5, ISBN 978-1-4020-5887-5, https://doi.org/10.1007/978-1-4020-5887-5_5, retrieved 2022-05-27
- ↑ 81.0 81.1 Tanz, Nicole; Schmidt, Hanns-Ludwig (2010-03-10). "δ 34 S-Value Measurements in Food Origin Assignments and Sulfur Isotope Fractionations in Plants and Animals" (in en). Journal of Agricultural and Food Chemistry 58 (5): 3139–3146. doi:10.1021/jf903251k. ISSN 0021-8561. PMID 20141140. https://pubs.acs.org/doi/10.1021/jf903251k.
- ↑ Jørgensen, Bo Barker (April 1982). "Mineralization of organic matter in the sea bed—the role of sulphate reduction". Nature 296 (5858): 643–645. doi:10.1038/296643a0. ISSN 0028-0836. http://dx.doi.org/10.1038/296643a0.
- ↑ 83.0 83.1 83.2 Cavallaro, Viviana; Maghrebi, Moez; Caschetto, Mariachiara; Sacchi, Gian Attilio; Nocito, Fabio Francesco (2022). "Sulfur Stable Isotope Discrimination in Rice: A Sulfur Isotope Mass Balance Study". Frontiers in Plant Science 13: 837517. doi:10.3389/fpls.2022.837517. ISSN 1664-462X. PMID 35360342.
- ↑ Sessions, Alex L.; Doughty, David M.; Welander, Paula V.; Summons, Roger E.; Newman, Dianne K. (2009-07-28). "The Continuing Puzzle of the Great Oxidation Event" (in en). Current Biology 19 (14): R567–R574. doi:10.1016/j.cub.2009.05.054. ISSN 0960-9822. PMID 19640495. http://www.sciencedirect.com/science/article/pii/S0960982209011890.
- ↑ Pavlov, A.a.; Kasting, J.f. (2002-03-01). "Mass-Independent Fractionation of Sulfur Isotopes in Archean Sediments: Strong Evidence for an Anoxic Archean Atmosphere". Astrobiology 2 (1): 27–41. doi:10.1089/153110702753621321. ISSN 1531-1074. PMID 12449853. Bibcode: 2002AsBio...2...27P.
- ↑ Zahnle, K.; Claire, M.; Catling, D. (2006). "The loss of mass-independent fractionation in sulfur due to a Palaeoproterozoic collapse of atmospheric methane" (in en). Geobiology 4 (4): 271–283. doi:10.1111/j.1472-4669.2006.00085.x. ISSN 1472-4669.
- ↑ Shen, Yanan; Farquhar, James; Masterson, Andrew; Kaufman, Alan J.; Buick, Roger (2009-03-30). "Evaluating the role of microbial sulfate reduction in the early Archean using quadruple isotope systematics" (in en). Earth and Planetary Science Letters 279 (3): 383–391. doi:10.1016/j.epsl.2009.01.018. ISSN 0012-821X. Bibcode: 2009E&PSL.279..383S. http://www.sciencedirect.com/science/article/pii/S0012821X09000296.
- ↑ Shen, Yanan; Buick, Roger (2004-02-01). "The antiquity of microbial sulfate reduction" (in en). Earth-Science Reviews 64 (3): 243–272. doi:10.1016/S0012-8252(03)00054-0. ISSN 0012-8252. Bibcode: 2004ESRv...64..243S. http://www.sciencedirect.com/science/article/pii/S0012825203000540.
- ↑ Canfield, Donald E.; Teske, Andreas (July 1996). "Late Proterozoic rise in atmospheric oxygen concentration inferred from phylogenetic and sulphur-isotope studies" (in en). Nature 382 (6587): 127–132. doi:10.1038/382127a0. ISSN 1476-4687. PMID 11536736. Bibcode: 1996Natur.382..127C. https://www.nature.com/articles/382127a0.
- ↑ Bontognali, Tomaso R. R.; Sessions, Alex L.; Allwood, Abigail C.; Fischer, Woodward W.; Grotzinger, John P.; Summons, Roger E.; Eiler, John M. (2012-09-18). "Sulfur isotopes of organic matter preserved in 3.45-billion-year-old stromatolites reveal microbial metabolism" (in en). Proceedings of the National Academy of Sciences 109 (38): 15146–15151. doi:10.1073/pnas.1207491109. ISSN 0027-8424. PMID 22949693.
- ↑ 91.0 91.1 91.2 Fike, D. A.; Grotzinger, J. P. (2008-06-01). "A paired sulfate–pyrite δ34S approach to understanding the evolution of the Ediacaran–Cambrian sulfur cycle" (in en). Geochimica et Cosmochimica Acta 72 (11): 2636–2648. doi:10.1016/j.gca.2008.03.021. ISSN 0016-7037. Bibcode: 2008GeCoA..72.2636F. http://www.sciencedirect.com/science/article/pii/S0016703708001749.
- ↑ 92.0 92.1 92.2 Raven, Morgan Reed; Fike, David A.; Bradley, Alexander S.; Gomes, Maya L.; Owens, Jeremy D.; Webb, Samuel A. (2019-04-15). "Paired organic matter and pyrite δ34S records reveal mechanisms of carbon, sulfur, and iron cycle disruption during Ocean Anoxic Event 2" (in en). Earth and Planetary Science Letters 512: 27–38. doi:10.1016/j.epsl.2019.01.048. ISSN 0012-821X. Bibcode: 2019E&PSL.512...27R.
- ↑ Owens, Jeremy D.; Gill, Benjamin C.; Jenkyns, Hugh C.; Bates, Steven M.; Severmann, Silke; Kuypers, Marcel M. M.; Woodfine, Richard G.; Lyons, Timothy W. (2013-11-12). "Sulfur isotopes track the global extent and dynamics of euxinia during Cretaceous Oceanic Anoxic Event 2" (in en). Proceedings of the National Academy of Sciences 110 (46): 18407–18412. doi:10.1073/pnas.1305304110. ISSN 0027-8424. PMID 24170863. Bibcode: 2013PNAS..11018407O.
- ↑ Pasquier, Virgil; Sansjofre, Pierre; Rabineau, Marina; Revillon, Sidonie; Houghton, Jennifer; Fike, David A. (2017-06-06). "Pyrite sulfur isotopes reveal glacial−interglacial environmental changes" (in en). Proceedings of the National Academy of Sciences 114 (23): 5941–5945. doi:10.1073/pnas.1618245114. ISSN 0027-8424. PMID 28533378. Bibcode: 2017PNAS..114.5941P.
- ↑ Fike, David A.; Bradley, Alexander S.; Rose, Catherine V. (2015-05-30). "Rethinking the Ancient Sulfur Cycle". Annual Review of Earth and Planetary Sciences 43 (1): 593–622. doi:10.1146/annurev-earth-060313-054802. ISSN 0084-6597. Bibcode: 2015AREPS..43..593F.
- ↑ 96.0 96.1 Kah, Linda C.; Lyons, Timothy W.; Frank, Tracy D. (October 2004). "Low marine sulphate and protracted oxygenation of the Proterozoic biosphere" (in en). Nature 431 (7010): 834–838. doi:10.1038/nature02974. ISSN 1476-4687. PMID 15483609. Bibcode: 2004Natur.431..834K. https://www.nature.com/articles/nature02974.
- ↑ Habicht, Kirsten S.; Gade, Michael; Thamdrup, Bo; Berg, Peter; Canfield, Donald E. (2002-12-20). "Calibration of Sulfate Levels in the Archean Ocean" (in en). Science 298 (5602): 2372–2374. doi:10.1126/science.1078265. ISSN 0036-8075. PMID 12493910. Bibcode: 2002Sci...298.2372H. https://www.science.org/doi/10.1126/science.1078265.
- ↑ Crowe, Sean A.; Paris, Guillaume; Katsev, Sergei; Jones, CarriAyne; Kim, Sang-Tae; Zerkle, Aubrey L.; Nomosatryo, Sulung; Fowle, David A. et al. (2014-11-07). "Sulfate was a trace constituent of Archean seawater" (in en). Science 346 (6210): 735–739. doi:10.1126/science.1258966. ISSN 0036-8075. PMID 25378621. Bibcode: 2014Sci...346..735C. https://www.science.org/doi/10.1126/science.1258966.
 |  |

