Chemistry:Arsenic biochemistry

Arsenic biochemistry refers to biochemical processes that can use arsenic or its compounds, such as arsenate. Arsenic is a moderately abundant element in Earth's crust, and although many arsenic compounds are often considered highly toxic to most life, a wide variety of organoarsenic compounds are produced biologically and various organic and inorganic arsenic compounds are metabolized by numerous organisms. This pattern is general for other related elements, including selenium, which can exhibit both beneficial and deleterious effects. Arsenic biochemistry has become topical since many toxic arsenic compounds are found in some aquifers,[1] potentially affecting many millions of people via biochemical processes.[2]
Sources of arsenic
Organoarsenic compounds in nature

The evidence that arsenic may be a beneficial nutrient at trace levels below the background to which living organisms are normally exposed has been reviewed.[3] Some organoarsenic compounds found in nature are arsenobetaine and arsenocholine,[4] both being found in many marine organisms.[2] Some As-containing nucleosides (sugar derivatives) are also known.[5] Several of these organoarsenic compounds arise via methylation processes. For example, the mold Scopulariopsis brevicaulis produces significant amounts of trimethylarsine if inorganic arsenic is present.[6] The organic compound arsenobetaine is found in some marine foods such as fish and algae, and also in mushrooms in larger concentrations. In clean environments, the edible mushroom species Cyanoboletus pulverulentus hyperaccumulates arsenic in concentrations reaching even 1,300 mg/kg in dry weight; cacodylic acid is the major As compound.[7] A very unusual composition of organoarsenic compounds was found in deer truffles (Elaphomyces spp.).[8] The average person's intake is about 10–50 µg/day. Values about 1000 µg are not unusual following consumption of fish or mushrooms; however, there is little danger in eating fish since this arsenic compound is nearly non-toxic.[9]
- Representative organoarsenic compounds found in nature.
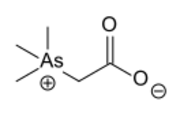
Arsenobetaine, one of the most common arsenic compound in nature. Also common is arsenocholine, which has CH2OH in place of CO2H).

Trimethylarsine, produced by microbial action on arsenate-derived pigments

Arsenic-containing ribose derivatives (R = several groups)



A topical source of arsenic are the green pigments once popular in wallpapers, e.g. Paris green. A variety of illness have been blamed on this compound, although its toxicity has been exaggerated.[10]
Trimethylarsine, once known as Gosio's gas, is an intensely malodorous organoarsenic compound that is commonly produced by microbial action on inorganic arsenic substrates.[11]
Arsenic (V) compounds are easily reduced to arsenic (III) and could have served as an electron acceptor on primordial Earth.[12] Lakes that contain a substantial amount of dissolved inorganic arsenic, harbor arsenic-tolerant biota.
Incorrect claims of arsenic-based life (phosphorus substitution)
Although phosphate and arsenate are structurally similar, there is no evidence that arsenic replaces phosphorus in DNA or RNA.[13] A 2010 experiment involving the bacteria GFAJ-1 that made this claim was refuted by 2012.[14][15]
Anthropogenic arsenic compounds
Anthropogenic (man-made) sources of arsenic, like the natural sources, are mainly arsenic oxides and the associated anions. Man-made sources of arsenic, include wastes from mineral processing, swine and poultry farms.[16] For example, many ores, especially sulfide minerals, are contaminated with arsenic, which is released in roasting (burning in air). In such processing, arsenide is converted to arsenic trioxide, which is volatile at high temperatures and is released into the atmosphere. Poultry and swine farms make heavy use of the organoarsenic compound roxarsone as an antibiotic in feed.[17][18] Some wood is treated with copper arsenates as a preservative. The mechanisms by which these sources affect "downstream" living organisms remains uncertain but are probably diverse. One commonly cited pathway involves methylation.[19]
The monomethylated acid, methanearsonic acid (CH3AsO(OH)2), is a precursor to fungicides (tradename Neoasozin) in the cultivation of rice and cotton. Derivatives of phenylarsonic acid (C6H5AsO(OH)2) are used as feed additives for livestock, including 4-hydroxy-3-nitrobenzenearsonic acid (3-NHPAA or Roxarsone), ureidophenylarsonic acid, and p-arsanilic acid. These applications are controversial as they introduce soluble forms of arsenic into the environment.
Arsenic-based drugs
Despite, or possibly because of, its long-known toxicity, arsenic-containing potions and drugs have a history in medicine and quackery that continues into the 21st century.[20][21] Starting in the early 19th century and continuing into the 20th century, Fowler's solution, a toxic concoction of sodium arsenite, was sold. The organoarsenic compound Salvarsan was the first synthetic chemotherapeutic agent, discovered by Paul Ehrlich.[21] The treatment, however, led to many problems causing long lasting health complications.[22] Around 1943 it was finally superseded by penicillin. The related drug Melarsoprol is still in use against late-state African trypanosomiasis (sleeping sickness), despite its high toxicity and possibly fatal side effects.
Arsenic trioxide (As2O3) inhibits cell growth and induces apoptosis (programmed cell death) in certain types of cancer cells,[23] which are normally immortal and can multiply without limit. In combination with all-trans retinoic acid, it is FDA-approved as first-line treatment for promyelocytic leukemia.
Methylation of arsenic
Inorganic arsenic and its compounds, upon entering the food chain, are progressively metabolised (detoxified) through a process of methylation.[19] The methylation occurs through alternating reductive and oxidative methylation reactions, that is, reduction of pentavalent to trivalent arsenic followed by addition of a methyl group (CH3).[24]

In mammals, methylation occurs in the liver by methyltransferases, the products being the (CH3)2AsOH (dimethylarsinous acid) and (CH3)2As(O)OH (dimethylarsinic acid), which have the oxidation states As(III) and As(V), respectively.[2] Although the mechanism of methylation of arsenic in humans has not been elucidated, the source of methyl is methionine, which suggests a role of S-adenosyl methionine.[25] Exposure to toxic doses begin when the liver's methylation capacity is exceeded or inhibited.
There are two major forms of arsenic that can enter the body, arsenic (III) and arsenic (V).[26] Arsenic (III) enters the cells though aquaporins 7 and 9, which is a type of aquaglyceroporin.[26] Arsenic (V) compounds use phosphate transporters to enter cells.[26] The arsenic (V) can be converted to arsenic (III) by the enzyme purine nucleoside phosphorylase.[26] This is classified as a bioactivation step, as although arsenic (III) is more toxic, it is more readily methylated.[27]
There are two routes by which inorganic arsenic compounds are methylated.[28] The first route uses Cyt19 arsenic methyltransferase to methylate arsenic (III) to a mono-methylated arsenic (V) compound.[26] This compound is then converted to a mono-methylated arsenic (III) compound using Glutathione S-Transferase Omega-1 (GSTO1).[26] The mono-methylated arsenic (V) compound can then be methylated again by Cyt19 arsenic methyltransferase, which forms a dimethyl arsenic (V) compound, which can be converted to a dimethyl arsenic (III) compound by Glutathione S-Transferase Omega-1 (GTSO1).[26] The other route uses glutathione (GSH) to conjugate with arsenic (III) to form an arsenic (GS) 3 complex.[26] This complex can form a monomethylated arsenic (III) GS complex, using Cyt19 arsenic methyltransferase, and this monomethylated GS complex is in equilibrium with the monomethylated arsenic (III).[26] Cyt19 arsenic methyltransferase can methylate the complex one more time, and this forms a dimethylated arsenic GS complex, which is in equilibrium with a dimethyl arsenic (III) complex.[26] Both of the mono-methylated and di-methylated arsenic compounds can readily be excreted in urine.[27] However, the monomethylated compound was shown to be more reactive and more toxic than the inorganic arsenic compounds to human hepatocytes (liver), keratinocytes in the skin, and bronchial epithelial cells (lungs).[29]
Studies in experimental animals and humans show that both inorganic arsenic and methylated metabolites cross the placenta to the fetus, however, there is evidence that methylation is increased during pregnancy and that it could be highly protective for the developing organism.[30]
Enzymatic methylation of arsenic is a detoxification process; it can be methylated to methylarsenite, dimethylarsenite or trimethylarsenite, all of which are trivalent. The methylation is catalyzed by arsenic methyltransferase (AS3MT) in mammals, which transfers a methyl group on the cofactor S-adenomethionine (SAM) to arsenic (III). An orthologue of AS3MT is found in bacteria and is called CmArsM. This enzyme was tested in three states (ligand free, arsenic (III) bound and SAM bound). Arsenic (III) binding sites usually use thiol groups of cysteine residues. The catalysis involves thiolates of Cys72, Cys174, and Cys224. In an SN2 reaction, the positive charge on the SAM sulfur atom pulls the bonding electron from the carbon of the methyl group, which interacts with the arsenic lone pair to form an As−C bond, leaving SAH.[31]
Excretion
In humans, the major route of excretion of most arsenic compounds is via the urine. The biological half-life of inorganic arsenic is about 4 days, but is slightly shorter following exposure to arsenate than to arsenite. The main metabolites excreted in the urine of humans exposed to inorganic arsenic are mono- and dimethylated arsenic acids, together with some unmetabolized inorganic arsenic.[25]
The biotransformation of arsenic for excretion is primarily done through the nuclear factor erythroid 2 related factor 2 (Nrf2) pathway.[32] Under normal conditions the Nrf2 is bound to Kelch-like ECH associated protein 1 (Keap1) in its inactive form.[33] With the uptake of arsenic within cells and the subsequent reactions that result in the production of reactive oxygen species (ROS), the Nrf2 unbinds and becomes active. Keap1 has reactive thiol moieties that bind ROS or electrophilic arsenic species such as monomethylted arsenic (III) and induces the release of Nrf2 which then travels through the cytoplasm to the nucleus.[34] The Nrf2 then activates antioxidant responsive element (ARE) as well as electrophilic responsive element (EpRE) both of which contribute in the increase of antioxidant proteins.[35] Of particular note in these antioxidant proteins is heme oxygenase 1 ([HO-1]), NAD(P)H-quinone oxidoreductase 1 (NQO1), and γ-glutamylcysteine synthase (γGCS) which work in conjunction to reduce the oxidative species such as hydrogen peroxide to decrease the oxidative stress upon the cell. The increase in γGCS causes an increased production of arsenite triglutathionine (As(SG)3) an important adduct that is taken up by either multidrug associated protein 1 or 2 (MRP1 or MRP2) which removes the arsenic out of the cell and into bile for excretion.[34] This adduct can also decompose back into inorganic arsenic.
Of particular note in the excretion of arsenic is the multiple methylation steps that take place which may increase the toxicity of arsenic[36] due to MMeAsIII being a potent inhibitor of glutathione peroxidase,[37] glutathione reductase, pyruvate dehydrogenase,[38] and thioredoxin reductase.[39]
Arsenic toxicity
Arsenic is a cause of mortality throughout the world; associated problems include heart, respiratory, gastrointestinal, liver, nervous and kidney diseases.[2][25]
Arsenic interferes with cellular longevity by allosteric inhibition of an essential metabolic enzyme pyruvate dehydrogenase (PDH) complex, which catalyzes the oxidation of pyruvate to acetyl-CoA by NAD+. With the enzyme inhibited, the energy system of the cell is disrupted resulting in a cellular apoptosis episode. Biochemically, arsenic prevents use of thiamine resulting in a clinical picture resembling thiamine deficiency. Poisoning with arsenic can raise lactate levels and lead to lactic acidosis.
Genotoxicity involves inhibition of DNA repair and DNA methylation. The carcinogenic effect of arsenic arises from the oxidative stress induced by arsenic. Arsenic's high toxicity naturally led to the development of a variety of arsenic compounds as chemical weapons, e.g. dimethylarsenic chloride. Some were employed as chemical warfare agents, especially in World War I. This threat led to many studies on antidotes and an expanded knowledge of the interaction of arsenic compounds with living organisms. One result was the development of antidotes such as British anti-Lewisite. Many such antidotes exploit the affinity of As(III) for thiolate ligands, which convert highly toxic organoarsenicals to less toxic derivatives. It is generally assumed that arsenates bind to cysteine residues in proteins.
By contrast, arsenic oxide is an approved and effective chemotherapeutic drug for the treatment of acute promyelocytic leukemia (APL).[3]
Toxicity of pentavalent arsenicals
Due to its similar structure and properties, pentavalent arsenic metabolites are capable of replacing the phosphate group of many metabolic pathways.[40] The replacement of phosphate by arsenate is initiated when arsenate reacts with glucose and gluconate in vitro.[40] This reaction generates glucose-6-arsenate and 6-arsenogluconate, which act as analogs for glucose-6-phosphate and 6-phosphogluconate.[40] At the substrate level, during glycolysis, glucose-6-arsenate binds as a substrate to glucose-6-phosphate dehydrogenase, and also inhibits hexokinase through negative feedback.[40] Unlike the importance of phosphate in glycolysis, the presence of arsenate restricts the generation of ATP by forming an unstable anhydride product, through the reaction with D-glyceraldehyde-3-phosphate.[40] The anhydride 1-arsenato-3-phospho-D-glycerate generated readily hydrolyzes due to the longer bond length of As-O compared to P-O.[40] At the mitochondrial level, arsenate uncouples the synthesis of ATP by binding to ADP in the presence of succinate, thus forming an unstable compound that ultimately results in a decrease of ATP net gain.[40] Arsenite (III) metabolites, on the other hand, have limited effect on ATP production in red blood cells.[40]
Toxicity of trivalent arsenicals
Enzymes and receptors that contain thiol or sulfhydryl functional groups are actively targeted by arsenite (III) metabolites.[40] These sulfur-containing compounds are normally glutathione and the amino acid cysteine.[40] Arsenite derivatives generally have higher binding affinity compared to the arsenate metabolites.[40] These bindings restrict activity of certain metabolic pathways.[40] For example, pyruvate dehydrogenase (PDH) is inhibited when monomethylarsonous acid (MMAIII) targets the thiol group of the lipoic acid cofactor.[40] PDH is a precursor of acetyl-CoA, thus the inhibition of PDH eventually limits the production of ATP in electron transport chain, as well as the production of gluconeogenesis intermediates.[40]
Oxidative stress
Arsenic can cause oxidative stress through the formation of reactive oxygen species (ROS), and reactive nitrogen species (RNS).[28] Reactive oxygen species are produced by the enzyme NADPH oxidase, which transfers electrons from NADPH to oxygen, synthesizing a superoxide, which is a reactive free radical. This superoxide can react to form hydrogen peroxide and a reactive oxygen species. The enzyme NADPH oxidase is able to generate more reactive oxygen species in the presence of arsenic, due to the subunit p22phax, which is responsible for the electron transfer, being upregulated by arsenic.[28] The reactive oxygen species are capable of stressing the endoplasmic reticulum, which increases the amount of the unfolded protein response signals.[28] This leads to inflammation, cell proliferation, and eventually to cell death.[28] Another mechanism in which reactive oxygen species cause cell death would be through the cytoskeleton rearrangement, which affects the contractile proteins.[28]
The reactive nitrogen species arise once the reactive oxygen species destroy the mitochondria.[28] This leads to the formation of the reactive nitrogen species, which are responsible for damaging DNA in arsenic poisoning.[28] Mitochondrial damage is known to cause the release of reactive nitrogen species, due to the reaction between superoxides and nitric oxide (NO).[28] Nitric oxide (NO) is a part of cell regulation, including cellular metabolism, growth, division and death.[28] Nitric oxide (NO) reacts with reactive oxygen species to form peroxynitrite.[28] In cases of chronic arsenic exposure, the nitric oxide levels are depleted, due to the superoxide reactions.[28] The enzyme NO synthase (NOS) uses L-arginine to form nitric oxide, but this enzyme is inhibited by monomethylated arsenic (III) compounds.[28]
DNA damage
Arsenic is reported to cause DNA modifications such as aneuploidy, micronuclei formation, chromosome abnormality, deletion mutations, sister chromatid exchange and crosslinking of DNA with proteins.[41] It has been demonstrated that arsenic does not directly interact with DNA and it is considered a poor mutagen, but instead, it helps mutagenicity of other carcinogens.[42] For instance, a synergistic increase in the mutagenic activity of arsenic with UV light has been observed in human and other mammalian cells after exposing the UV-treated cells to arsenic.[43][44] A series of experimental observations suggest that the arsenic genotoxicity is primarily linked to the generation of reactive oxygen species (ROS) during its biotransformation.[45][46][47] The ROS production is able to generate DNA adducts, DNA strand breaks, crosslinks and chromosomal aberrations.[48][49][50][51] The oxidative damage is caused by modification of DNA nucleobases, in particular 8-oxoguanine (8-OHdG) which leads to G:C to T:A mutations.[52] Inorganic arsenic can also cause DNA strand break even at low concentrations.[53]
Inhibition of DNA repair
Inhibition of DNA repair processes is considered one of main mechanism of inorganic arsenic genotoxicity. Nucleotide excision repair (NER) and base excision repair (BER) are the processes implicated in the repair of DNA base damage induced by ROS after arsenic exposure. In particular, the NER mechanism is the major pathway for repairing bulky distortions in DNA double helix, while the BER mechanism is mainly implicated in the repair of single strand breaks induced by ROS,[54][55][56][57] but inorganic arsenic could also repress the BER mechanism.[58][59][60]
Neurodegenerative mechanisms
Arsenic is highly detrimental to the innate and the adaptive immune system of the body.[61] When the amount of unfolded and misfolded proteins in endoplasmic reticulum stress is excessive, the unfolded protein response (UPR) is activated to increase the activity of several receptors that are responsible the restoration of homeostasis.[61] The inositol-requiring enzyme-1 (IRE1) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) are two receptors that restrict the rate of translation.[61] On the other hand, the unfolded proteins are corrected by the production of chaperones, which are induced by the activating transcription factor 6 (ATF6).[61] If the number of erroneous proteins elevates, further mechanism is active which triggers apoptosis.[61] Arsenic has evidentially shown to increase the activity of these protein sensors.[61]
Immune dysfunction
Arsenic exposure in small children distorts the ratio of T helper cells (CD4) to cytotoxic T cells (CD8), which are responsible for immunodepression.[62] In addition, arsenic also increases the number of inflammatory molecules being secreted through macrophages.[62] The excess amount of granulocytes and monocytes lead to a chronic state of inflammation, which might result in cancer development.[62]
Arsenic poisoning treatment
There are three molecules that serve as chelator agents that bond to arsenic. These three are British Anti-Lewisite (BAL, Dimercaprol), succimer (DMSA) and Unithiol (DMPS).[63]
When these agents chelate inorganic arsenic, it is converted into an organic form of arsenic because it is bound to the organic chelating agent. The sulfur atoms of the thiol groups are the site of interaction with arsenic. This is because the thiol groups are nucleophilic while the arsenic atoms are electrophilic. Once bound to the chelating agent the molecules can be excreted, and therefore free inorganic arsenic atoms are removed from the body.
Other chelating agents can be used, but may cause more side effects than British Anti-Lewisite (BAL, Dimercaprol), succimer (DMSA) and (DMPS). DMPS and DMSA also have a higher therapeutic index than BAL.[63]
These drugs are efficient for acute poisoning of arsenic, which refers to the instantaneous effects caused by arsenic poisoning. For example, headaches, vomiting or sweating are some of the common examples of an instantaneous effect. In comparison, chronic poisonous effects arise later on, and unexpectedly such as organ damage. Usually it is too late to prevent them once they appear. Therefore, action should be taken as soon as acute poisonous effects arise.[64]
See also
- Arsenic compounds
- Extremophile
- Geomicrobiology
- Hypothetical types of biochemistry
- Organoarsenic chemistry
References
- ↑ Pearce, Fred (2006). When the Rivers Run Dry: Journeys Into the Heart of the World's Water Crisis. Toronto: Key Porter. ISBN 978-1-55263-741-8.
- ↑ 2.0 2.1 2.2 2.3 Elke Dopp, Andrew D. Kligerman and Roland A. Diaz-Bone Organoarsenicals. Uptake, Metabolism, and Toxicity 2010, Royal Society of Chemistry. ISBN:978-1-84973-082-2. doi:10.1039/9781849730822-00231
- ↑ 3.0 3.1 Wilcox, Dean E. (2013). "Arsenic. Can This Toxic Metalloid Sustain Life?". in Astrid Sigel, Helmut Sigel and Roland K. O. Sigel. Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. 13. Springer. pp. 475–498. doi:10.1007/978-94-007-7500-8_15. ISBN 978-94-007-7499-5.
- ↑ Arsenocholine - Structure and Data
- ↑ Francesconi, Kevin A.; Edmonds, John S.; Stick, Robert V. (1992). "Arsenic compounds from the kidney of the giant clam Tridacna maxima: Isolation and identification of an arsenic-containing nucleoside". Journal of the Chemical Society, Perkin Transactions 1 (11): 1349. doi:10.1039/P19920001349.
- ↑ Bentley, Ronald; Chasteen, TG (2002). "Microbial Methylation of Metalloids: Arsenic, Antimony, and Bismuth". Microbiology and Molecular Biology Reviews 66 (2): 250–271. doi:10.1128/MMBR.66.2.250-271.2002. PMID 12040126.
- ↑ Braeuer, Simone; Goessler, Walter; Kameník, Jan; Konvalinková, Tereza; Žigová, Anna; Borovička, Jan (2018). "Arsenic hyperaccumulation and speciation in the edible ink stain bolete ( Cyanoboletus pulverulentus )". Food Chemistry 242: 225–231. doi:10.1016/j.foodchem.2017.09.038. PMID 29037683.
- ↑ Braeuer, Simone; Borovička, Jan; Goessler, Walter (2018-02-12). "A unique arsenic speciation profile in Elaphomyces spp. ("deer truffles")—trimethylarsine oxide and methylarsonous acid as significant arsenic compounds" (in en). Analytical and Bioanalytical Chemistry 410 (9): 2283–2290. doi:10.1007/s00216-018-0903-3. ISSN 1618-2642. PMID 29430602.
- ↑ Cullen, William R; Reimer, Kenneth J. (1989). "Arsenic speciation in the environment". Chemical Reviews 89 (4): 713–764. doi:10.1021/cr00094a002. https://atrium.lib.uoguelph.ca/xmlui/bitstream/10214/2162/1/i6_le.pdf.
- ↑ Ronald Bentley and Thomas G. Chasteen (2002). "Microbial Methylation of Metalloids: Arsenic, Antimony, and Bismuth". Microbiology and Molecular Biology Reviews 66 (2): 250–271. doi:10.1128/MMBR.66.2.250-271.2002. PMID 12040126.
- ↑ Cullen, William R.; Reimer, Kenneth J. (1989). "Arsenic speciation in the environment". Chemical Reviews 89 (4): 713–764. doi:10.1021/cr00094a002.
- ↑ Oremland, Ronald S.; Saltikov, Chad W.; Wolfe-Simon, Felisa; Stolz, John F. (2009). "Arsenic in the Evolution of Earth and Extraterrestrial Ecosystems". Geomicrobiology Journal 26 (7): 522–536. doi:10.1080/01490450903102525.
- ↑ Westheimer, F.H. (6 June 1987). "Why nature chose phosphates". Science 235 (4793): 1173–1178 (see pp. 1175–1176). doi:10.1126/science.2434996. PMID 2434996. Bibcode: 1987Sci...235.1173W.
- ↑ Erb, T. J.; Kiefer, P.; Hattendorf, B.; Gunther, D.; Vorholt, J. A. (2012). "GFAJ-1 is an Arsenate-Resistant, Phosphate-Dependent Organism". Science 337 (6093): 467–70. doi:10.1126/science.1218455. PMID 22773139. Bibcode: 2012Sci...337..467E.
- ↑ Reaves, M. L.; Sinha, S.; Rabinowitz, J. D.; Kruglyak, L.; Redfield, R. J. (2012). "Absence of Detectable Arsenate in DNA from Arsenate-Grown GFAJ-1 Cells". Science 337 (6093): 470–3. doi:10.1126/science.1219861. PMID 22773140. Bibcode: 2012Sci...337..470R.
- ↑ Nordstrom DK (2002). "Worldwide occurrences of arsenic in ground water". Science 296 (5576): 2143–2145. doi:10.1126/science.1072375. PMID 12077387.
- ↑ Hileman, B (9 April 2007). "Arsenic in Chicken Production". Chemical and Engineering News. pp. 34–35.
- ↑ Bottemiller, Helena (26 September 2009). "Bill Introduced to Ban Arsenic Antibiotics in Feed". Food Safety News. http://www.foodsafetynews.com/2009/09/bill-introduced-to-ban-arsenic-antibiotics-in-feed/.
- ↑ 19.0 19.1 Sakurai T (2003). "Biomethylation of Arsenic is Essentially Detoxicating Event". Journal of Health Science 49 (3): 171–178. doi:10.1248/jhs.49.171. http://www.jstage.jst.go.jp/article/jhs/49/3/49_171/_article/-char/en. Retrieved 2011-01-10.
- ↑ Jun Zhu; Zhu Chen; Valérie Lallemand-Breitenbach; Hugues de Thé (2002). "How Acute Promyelocytic Leukaemia Revived Arsenic". Nature Reviews Cancer 2 (9): 705–714. doi:10.1038/nrc887. PMID 12209159.
- ↑ 21.0 21.1 Gibaud, Stéphane; Jaouen, Gérard (2010). "Arsenic-Based Drugs: From Fowler's Solution to Modern Anticancer Chemotherapy". Medicinal Organometallic Chemistry. Topics in Organometallic Chemistry. 32. pp. 1–20. doi:10.1007/978-3-642-13185-1_1. ISBN 978-3-642-13184-4. Bibcode: 2010moc..book....1G.
- ↑ Elschenbroich, C. ”Organometallics” (2006) Wiley-VCH: Weinheim. ISBN:978-3-527-29390-2
- ↑ Park, Woo H. Park; Jae G. Seol; Eun S. Kim; Jung M. Hyun; Chul W. Jung; Chung C. Lee; Byoung K. Kim; Young Y. Lee (June 6, 2000). "Arsenic Trioxide-mediated Growth Inhibition in MC/CAR Myeloma Cells via Cell Cycle Arrest in Association with Induction of Cyclin-dependent Kinase Inhibitor, p21, and Apoptosis". Cancer Research 60 (3065): 3065–71. PMID 10850458. http://cancerres.aacrjournals.org/content/60/11/3065.full. Retrieved 2010-12-15.
- ↑ "Arsenic in Drinking Water - Review article". IARC Monographs 84: 133–135. http://monographs.iarc.fr/ENG/Monographs/vol84/mono84-6.pdf. Retrieved 2011-01-10.
- ↑ 25.0 25.1 25.2 "Arsenic in Drinking Water - Review article". IARC Monographs - World Health Organization 84. http://monographs.iarc.fr/ENG/Monographs/vol84/mono84-6.pdf. Retrieved 2011-01-10.
- ↑ 26.0 26.1 26.2 26.3 26.4 26.5 26.6 26.7 26.8 26.9 Kumagai, Yoshito; Sumi, Daigo (2007). "Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity". Annual Review of Pharmacology and Toxicology 47: 243–62. doi:10.1146/annurev.pharmtox.47.120505.105144. PMID 17002598.
- ↑ 27.0 27.1 Vahter, Marie (2002). "Mechanisms of arsenic biotransformation". Toxicology 181–182: 211–7. doi:10.1016/S0300-483X(02)00285-8. PMID 12505313.
- ↑ 28.00 28.01 28.02 28.03 28.04 28.05 28.06 28.07 28.08 28.09 28.10 28.11 28.12 Hunt, Katherine M.; Srivastava, Ritesh K.; Elmets, Craig A.; Athar, Mohammad (2014). "The mechanistic basis of arsenicosis: Pathogenesis of skin cancer". Cancer Letters 354 (2): 211–9. doi:10.1016/j.canlet.2014.08.016. PMID 25173797.
- ↑ Petrick, Jay S.; Ayala-Fierro, Felix; Cullen, William R.; Carter, Dean E.; Vasken Aposhian, H. (2000). "Monomethylarsonous Acid (MMAIII) is More Toxic Than Arsenite in Chang Human Hepatocytes". Toxicology and Applied Pharmacology 163 (2): 203–7. doi:10.1006/taap.1999.8872. PMID 10698679.
- ↑ "Arsenic in Drinking Water - Review article". IARC Monographs 84: 138. http://monographs.iarc.fr/ENG/Monographs/vol84/mono84-6.pdf. Retrieved 2011-01-10.
- ↑ Ajees, A.A. (July 10, 2012). "Structure of an As(III) S-Adenosylmethionine Methyltransferase: insights into the Mechanism of Arsenic Biotransformation". Biochemistry 51 (27): 5476–5485. doi:10.1021/bi3004632. PMID 22712827.
- ↑ Kumagai, Yoshito; Sumi, Daigo Sumi (2007). "Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity". Annual Review of Pharmacology and Toxicology 47: 243–62. doi:10.1146/annurev.pharmtox.47.120505.105144. PMID 17002598.
- ↑ Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J. D.; Yamamoto, M (1999). "Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain". Genes Dev. 13 (1): 76–86. doi:10.1101/gad.13.1.76. PMID 9887101.
- ↑ 34.0 34.1 Kumagai, Yoshito; Sumi, Daigo Sumi (2007). "Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity". Annual Review of Pharmacology and Toxicology 47: 243–62. doi:10.1146/annurev.pharmtox.47.120505.105144. PMID 17002598.
- ↑ Pi, J; Waalkes, MP; Kumagai, Y; Reece, JM; Qu, W (2003). "Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide". Exp. Cell Res. 290 (2): 234–45. doi:10.1016/s0014-4827(03)00341-0. PMID 14567983. https://zenodo.org/record/1259581.
- ↑ Stýblo, M.; Drobná, Z.; Jaspers, I.; Lin, S.; Thomas, D. J. (2002). "The role of biomethylation in toxicity and carcinogenicity of arsenic: A research update". Environmental Health Perspectives 110 (Suppl 5): 767–771. doi:10.1289/ehp.110-1241242. PMID 12426129.
- ↑ Chouchane, S.; Snow, E. T.; Snow, E. T. (2001). "In vitro effect of arsenical compounds on glutathione-related enzymes". Chem. Res. Toxicol. 14 (5): 517–22. doi:10.1021/tx000123x. PMID 11368549.
- ↑ Petrick, Jay S.; Jagadish, Bhumasamudram; Mash, Eugene A.; Aposhian, H. Vasken (2001). "Monomethylarsonous Acid (MMAIII) and Arsenite: LD50in Hamsters and in Vitro Inhibition of Pyruvate Dehydrogenase". Chemical Research in Toxicology 14 (6): 651–656. doi:10.1021/tx000264z. PMID 11409934.
- ↑ Lin, Lin S.; Thomas, D. J.; Cullen, W. R.; Wang, C.; Styblo, M.; Del Razo, L. M. (2001). "Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes". Chem. Res. Toxicol. 14 (3): 305–11. doi:10.1021/tx0001878. PMID 11258980.
- ↑ 40.00 40.01 40.02 40.03 40.04 40.05 40.06 40.07 40.08 40.09 40.10 40.11 40.12 40.13 Hughes, Michael F (2002). "Arsenic toxicity and potential mechanisms of action". Toxicology Letters 133 (1): 1–16. doi:10.1016/S0378-4274(02)00084-X. PMID 12076506. https://zenodo.org/record/1260065.
- ↑ Rossman, T.G (2003). "Mechanism of arsenic carcinogenesis: An integrated approach". Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 533 (1–2): 37–65. doi:10.1016/j.mrfmmm.2003.07.009. PMID 14643412.
- ↑ Pierce, B.L; Kibriya, M.G (2012). "Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh". PLOS Genetics 8 (2): e1002522. doi:10.1371/journal.pgen.1002522. PMID 22383894.
- ↑ Li, J.H; Rossman, T.G (1991). "Comutagenesis of sodium arsenite with ultraviolet radiation in Chinese hamster V79 cells". Biology of Metals 4 (4): 197–200. doi:10.1007/BF01141180. PMID 1777354.
- ↑ Lee, T.C; Oshimura, M (1985). "Comparison of arsenic-induced cell transformation, cytotoxicity, mutation and cytogenetic effects in Syrian hamster embryo cells in culture". Carcinogenesis 6 (10): 1421–1426. doi:10.1093/carcin/6.10.1421. PMID 3840060. https://zenodo.org/record/1234307.
- ↑ Kessel, M; Liu, S.X (2002). "Arsenic induces oxidative DNA damage in mammalian cells". Molecular and Cellular Biochemistry 234/235 (1–2): 234–235:301–308. doi:10.1023/A:1015927406142. PMID 12162448.
- ↑ Nesnow, S; Roop, B.C (2002). "DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species". Chemical Research in Toxicology 15 (12): 1627–1634. doi:10.1021/tx025598y. PMID 12482246.
- ↑ Jomova, K; Jenisova, Z (2011). "Arsenic: Toxicity, oxidative stress and human disease". Journal of Applied Toxicology 31 (2): 95–107. doi:10.1002/jat.1649. PMID 21321970.
- ↑ Kitchin, K.T; Wallace, K (2008). "Evidence against the nuclear in situ binding of arsenicals—Oxidative stress theory of arsenic carcinogenesis". Toxicology and Applied Pharmacology 232 (2): 252–257. doi:10.1016/j.taap.2008.06.021. PMID 18671993.
- ↑ Bau, D.T; Wang, T.S (2002). "Oxidative DNA adducts and DNA-protein cross-links are the major DNA lesions induced by arsenite". Environmental Health Perspectives 110 (Suppl 5): 753–756. doi:10.1289/ehp.02110s5753. PMID 12426126.
- ↑ Hwang, E.S; Kim, G.H (2007). "Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research". Toxicology 229 (1–2): 1–10. doi:10.1016/j.tox.2006.10.013. PMID 17118505.
- ↑ Liu, Su X. (December 2000). "Induction of oxyradicals by arsenic: Implication for mechanism of genotoxicity". Proceedings of the National Academy of Sciences of the United States of America 98 (4): 1643–1648. doi:10.1073/pnas.98.4.1643. PMID 11172004. Bibcode: 2001PNAS...98.1643L.
- ↑ Grollman, A.P; Moriya, M (1993). "Mutagenesis by 8-oxoguanine: An enemy within". Trends in Genetics 9 (7): 246–249. doi:10.1016/0168-9525(93)90089-Z. PMID 8379000.
- ↑ Martinez, V.D; Vucic, E.A (2011). "Arsenic biotransformation as a cancer promoting factor by inducing DNA damage and disruption of repair mechanisms". Molecular Biology International 2011: 718974. doi:10.4061/2011/718974. PMID 22091411.
- ↑ Lai, Y; Zhao, W (2011). "Role of DNA polymerase beta in the genotoxicity of arsenic". Environmental and Molecular Mutagenesis 52 (6): 460–468. doi:10.1002/em.20643. PMID 21370284.
- ↑ Hartwig, A; Groblinghoff, U.D (1997). "Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts". Carcinogenesis 18 (2): 399–405. doi:10.1093/carcin/18.2.399. PMID 9054635.
- ↑ Curnow, A; Salter, L (2001). "A preliminary investigation of the effects of arsenate on irradiation-induced DNA damage in cultured human lung fibroblasts". Journal of Toxicology and Environmental Health, Part A 63 (8): 605–616. doi:10.1080/152873901316857789. PMID 11549120.
- ↑ Schwerdtle, T; Walter, I (2003). "Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA". Carcinogenesis 24 (5): 967–974. doi:10.1093/carcin/bgg018. PMID 12771042.
- ↑ Lai, Y; Zhao, W (2011). "Role of DNA polymerase beta in the genotoxicity of arsenic". Environmental and Molecular Mutagenesis 52 (6): 460–468. doi:10.1002/em.20643. PMID 21370284.
- ↑ Ebert, F; Weiss, A (2011). "Arsenicals affect base excision repair by several mechanisms". Mutat. Res. 715 (1–2): 32–41. doi:10.1016/j.mrfmmm.2011.07.004. PMID 21782832.
- ↑ Sykora, P; Snow, E.T (2008). "Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite". Toxicology and Applied Pharmacology 228 (3): 385–394. doi:10.1016/j.taap.2007.12.019. PMID 18252256. https://figshare.com/articles/journal_contribution/22865108.
- ↑ 61.0 61.1 61.2 61.3 61.4 61.5 Hunt, K. M; Srivastava, R. K; Elmets, C. A; Athar, M. (2014). "The mechanistic basis of arsenicosis: Pathogenesis of skin cancer". Cancer Letters 354 (2): 211–219. doi:10.1016/j.canlet.2014.08.016. PMID 25173797.
- ↑ 62.0 62.1 62.2 Vega, L. Environmental Health Risks. Nova Science Publishers. pp157-159. ISBN:978-1-60741-781-1
- ↑ 63.0 63.1 Kosnett, M. J. (2013). "The Role of Chelation in the Treatment of arsenic and Mercury Poisoning". Journal of Medical Toxicology 9 (4): 347–357. doi:10.1007/s13181-013-0344-5. PMID 24178900.
- ↑ "Acute & Chronic Poisoning Affects". http://www.medtox.org.uk/acute-chronic-poisoning-affects.html.
 |  |

