Chemistry:Migratory insertion
In organometallic chemistry, a migratory insertion is a type of reaction wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:[1]
Overview
In the migratory insertion, a ligand that is viewed as an anion (X) ligand in and a ligand that is viewed as neutral couple, generating a new anionic ligand. The anion and neutral ligands that react are adjacent. If the precursor complex is coordinatively saturated, migratory insertion often result in a coordinatively unsaturated product. A new (neutral) ligand can then react with the metal leading to a further insertion. The process can occur many times on a single metal, as in olefin polymerization.
The anionic ligand can be: H− (hydride), R− (alkyl), acyl, Ar− (aryl), or OR− (alkoxide). The ability of these groups to migrate is called their migratory aptitude. The neutral ligand can be CO, alkene, alkyne, or in some cases, even carbene.
Diverse reactions apply to the migratory insertion. One mechanism involves the attack of the anionic ligand on the electrophilic part of the neutral ligand (the anionic ligand migrates to the neutral ligand). The other mechanism involves the neutral ligand that inserts itself between the metal and the anionic ligand.
CO insertion
The insertion of carbon monoxide into a metal-carbon bond to form an acyl group is the basis of carbonylation reactions, which provides many commercially useful products. Mechanistic. Studies reveal that the alkyl group migrates intramolecularly to an adjacent CO ligand.[2][3]

CO Insertion reaction pathway for an octahedral complex

Early studies were conducted on the conversion of CH
3Mn(CO)
5 to give the acetyl derivative.[4] Using 13CO, the products is cis [Mn(COCH3)(13CO)(CO)4] (scheme 1).

CO insertion does not always involve migration. Treatment of CpFe(L)(CO)CH3 with 13CO yields a mix of both alkyl migration products and products formed by true insertion of bound carbonyls into the methyl group. Product distribution is influenced by the choice of solvent.[5]
Alkyl derivatives of square planar complexes undergo CO insertions particularly readily. Insertion reactions on square planar complexes are of particular interest because of their industrial applications. Since square planar complexes are often coordinatively unsaturated, they are susceptible to formation of 5-coordinate adducts, which undergo migratory insertion readily.[5] In most cases the in-plane migration pathway is preferred, but, unlike the nucleophilic pathway, is inhibited by an excess of CO.[6]

Effects on reaction rates
- Steric effects strain – Increasing the steric strain of the chelate backbone in square planar complexes pushes the carbonyl and methyl groups closer together, increasing the reactivity of insertion reactions.[6]
- Oxidation state – Oxidation of the metal tends to increase insertion reaction rates. The main rate-limiting step in the mechanism is the migration of the methyl group onto a carbonyl ligand, oxidizing the metal by imparting a greater partial positive charge on the acetyl carbon, and thus increasing the rate of reaction.[7]
- Lewis acids – Lewis acids also increase the reaction rates, for reasons similar to metal oxidation increasing the positive charge on the carbon. Lewis acids bind to the CO oxygen and remove charge, increasing the electrophilicity of the carbon. This can increase the reaction rate by a factor of up to 108, and the complex formed is stable enough that the reaction proceeds even without additional CO to bind to the metal.[7]
- Electronegativity of the leaving group - Increasing the electronegativity of the leaving alkyl group stabilizes the metal-carbon bond interaction and thus increases the activation energy required for migration, decreasing the reaction rate.[8]
- Trans-effect – Ligands in an octahedral or square planar complex are known to influence the reactivity of the group to which they are trans. This ligand influence is often referred to as the trans-influence, and it varies in intensity between ligands. A partial list of trans-influencing ligands is as follows, from highest trans-effect to lowest:[5] aryl, alkyl > NR3 > PR3 > AsR3 > CO > Cl. Ligands with a greater trans-influence impart greater electrophilicity to the active site. Increasing the electrophilicity of the CO group has been shown experimentally to greatly increase the reaction rate, while decreasing the electrophilicity of the methyl group slightly increases the reaction rate. This can be demonstrated by reacting a square planar [(PN)M(CO)(CH3)] complex with CO, where PN is a bidentate phosphorus- or nitrogen-bound ligand. This reaction proceeds in much greater yield when the methyl group is trans-P and the CO trans-N, owing to the higher trans-influence of the more electronegative nitrogen.[6]
Reverse reaction
Decarbonylation of aldehydes, the reverse of CO insertion, is a well-recognized reaction:
- RCHO → RH + CO
The reaction is not widely practiced in part because the alkanes are less useful materials than are the aldehyde precursors. Furthermore, the reaction is not often conducted catalytically because the extruded CO can be slow to dissociate.[9] Extrusion of CO from an organic aldehyde is most famously demonstrated using Wilkinson's catalyst:[10]
- RhCl(PPh3)3 + RCHO → RhCl(CO)(PPh3)2 + RH + PPh3
Please see Tsuji-Wilkinson Decarbonylation Reaction for an example of this elementary organometallic step in synthesis
Insertion of other oxides
Many electrophilic oxides insert into metal carbon bonds that include sulfur dioxide, carbon dioxide, and nitric oxide. These reactions have limited or no practical significance, but are of historic interest. With transition metal alkyls, these oxides behave as electrophiles and insert into the bond between metals and their relatively nucleophilic alkyl ligands. As discussed in the article on Metal sulfur dioxide complexes, the insertion of SO2 has been examined in particular detail. SO2 inserts to give both O-sulphinates and S-sulphinates, depending on the metal centre.[11] With square planar alkyl complexes, a pre-equilibrium is assumed involving formation of an adduct.[12]
Insertion of alkenes into metal-carbon bonds
The insertion of alkenes into both metal-carbon is important. The insertion of ethylene and propylene into titanium alkyls is the cornerstone of Ziegler–Natta catalysis, the main source of polyethylene and polypropylene. The majority of this technology involves heterogeneous catalysts, but it is widely assumed that the principles and observations on homogeneous systems are applicable to the solid-state versions. Related technologies include the Shell Higher Olefin Process which produces detergent precursors.

Steps in alkene polymerization. Step i involves binding of the monomer to the metal and step ii involves the migratory insertion step. These steps, which alternate from one side of the metal center to the other, are repeated many times for each polymer chain. The box represents a vacant (or extremely labile) coordination site.

Mechanism
Factors affecting the rate of olefin insertions include the formation of the cyclic, planar, four-center transition state involving incipient formation of a bond between the metal and an olefin carbon. From this transition state, it can be seen that a partial positive charge forms on the β-carbon with a partial negative charge formed on the carbon initially bonded to the metal. This polarization explains the subsequently observed formation of the bond between the negatively charged carbon/hydrogen and the positively charged β-carbon as well as the simultaneous formation of the metal-α-carbon bond. This transition state also highlights the two factors that most strongly contribute to the rate of olefin insertion reactions: (i) orbital overlap of the alkyl group initially attached to the metal and (ii) the strength of the metal-alkyl bond. With greater orbital overlap between the partially positive β-carbon and the partially negative hydrogen/alkyl group carbon, the formation of the new C-C bond is facilitated. With increasing strength of the metal-alkyl bond, the breaking of the bond between the metal and the hydrogen/alkyl carbon bond to form the two new bonds with the α-carbon and β-carbon (respectively) is slower, thus decreasing the rate of the insertion reaction.[13]
Insertion of alkenes into M–H bonds
The insertion of alkenes into metal-hydrogen bonds is a key step in hydrogenation and hydroformylation reactions. The reaction involves the alkene and the hydride ligands combining within the coordination sphere of a catalyst. In hydrogenation, the resulting alkyl ligand combines with a second hydride to give the alkane. Analogous reactions apply to the hydrogenation of alkynes: an alkenyl ligand combines with a hydride to eliminate an alkene.

Mechanism
In terms of mechanism, the insertion of alkenes into M–H bond and into M–C bonds are described similarly. Both involve four-membered transition states that place the less substituted carbon on the metal.
The reverse of olefin insertion into a metal-hydrogen bond is β-hydride elimination. The Principle of Microscopic Reversibility requires that the mechanism of β-hydride elimination follow the same pathway as the insertion of alkenes into metal hydride bonds. The first requirement for β-hydride elimination is the presence of a hydrogen at a position that is β with respect to the metal. β-elimination requires a vacant coordination position on the metal that will accommodate the hydrogen that is abstracted.[14]
Industrial applications
Carbonylation
Two widely employed applications of migratory insertion of carbonyl groups are hydroformylation and the production of acetic acid by carbonylation of methanol. The former converts alkenes, hydrogen, and carbon monoxide into aldehydes. The production of acetic acid by carbonylation proceeds via two similar industrial processes. More traditional is the Monsanto acetic acid process, which relies on a rhodium-iodine catalyst to transform methanol into acetic acid. This process has been superseded by the Cativa process which uses a related iridium catalyst, [Ir(CO)2I2]− (1).[15][16] By 2002, worldwide annual production of acetic acid stood at 6 million tons, of which approximately 60% is produced by the Cativa process.[15]
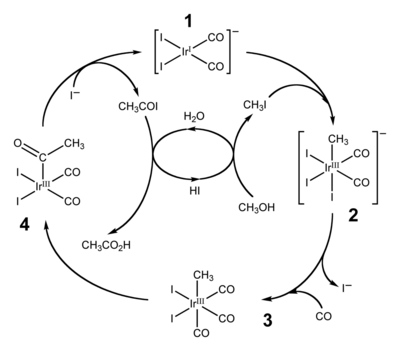
The catalytic cycle of the Cativa process

The Cativa process catalytic cycle, shown above, includes both insertion and de-insertion steps. The oxidative addition reaction of methyl iodide with (1) involves the formal insertion of the iridium(I) centre into the carbon-iodine bond, whilst step (3) to (4) is an example of migratory insertion of carbon monoxide into the iridium-carbon bond. The active catalyst species is regenerated by the reductive elimination of acetyl iodide from (4), a de-insertion reaction.[15]
Alkene polymerization
Industrial applications of alkene insertions include metal-catalyzed routes to polyethylene and polypropylene. Typically these conversions are catalyzed heterogeneously by titanium trichloride which are activated by aluminium alkyls. This technology is known as Ziegler–Natta catalysts.[17] In these reactions, ethylene coordinates to titanium metal followed by its insertion. These steps can be repeated multiple times, potentially leading to high molecular weight polymers.
References
- ↑ Elschenbroich, C. (2006). Organometallics. Weinheim: Wiley-VCH. ISBN 978-3-527-29390-2.
- ↑ Hartwig, J. F. (2010). Organotransition Metal Chemistry, from Bonding to Catalysis. New York, NY: University Science Books. ISBN 978-1-891389-53-5.
- ↑ Yadav, M. S. (2005). Quick Review in Inorganic Chemistry. Anmol Publications. p. 244. ISBN 978-81-261-1898-4. https://books.google.com/books?id=qlwlWsaoLwcC&q=Mn%28CO%295CH3+insertion+reaction&pg=PA244.
- ↑ F. Calderazzo, F. A. Cotton. “The Carbonylation of Methyl Manganese Pentacarbonyl and Decarbonylation of Acetyl Manganese Pentacarbonyl”. Inorg. Chem., 1962, 1, 30–36. https://doi.org/10.1021/ic50001a008.
- ↑ 5.0 5.1 5.2 Anderson, G. K.; Cross, R. J. (1984). "Carbonyl-Insertion Reactions of Square Planar Complexes". Acc. Chem. Res. 17 (2): 67–74. doi:10.1021/ar00098a005.
- ↑ 6.0 6.1 6.2 Cavell, K. J. (1996). "Recent Fundamental Studies on Migratory Insertion into Metal-Carbon Bonds". Coord. Chem. Rev. 155 (11): 209–243. doi:10.1016/S0010-8545(96)90182-4.
- ↑ 7.0 7.1 Alexander, J.J. (1985). "Insertions into transition metal-carbon bonds". The chemistry of the metal–carbon bond. 2. John Wiley & Sons. pp. 339–400. doi:10.1002/9780470771747.ch5. ISBN 978-0-470-77174-7.
- ↑ Shusterman, A. J.; Tamir, I.; Pross, A. (1988). "The Mechanism of Organometallic Migration Reactions. A Configuration Mixing (CM)Approach". J. Organomet. Chem. 340 (2): 203–222. doi:10.1016/0022-328X(88)80076-7.
- ↑ Fristrup, Peter; Kreis, Michael; Palmelund, Anders; Norrby, Per-Ola; Madsen, Robert (2008). "The Mechanism for the Rhodium-Catalyzed Decarbonylation of Aldehydes: A Combined Experimental and Theoretical Study". J. Am. Chem. Soc. 130 (15): 5206–5215. doi:10.1021/ja710270j. PMID 18303836. Bibcode: 2008JAChS.130.5206F.
- ↑ Ohno, K.; Tsuji, J. (1968). "Organic Synthesis by means of Noble Metal Compounds. XXXV. Novel Decarbonylation Reactions of Aldehydes and Acyl Halides using Rhodium Complexes". J. Am. Chem. Soc. 90 (1): 99–107. doi:10.1021/ja01003a018. Bibcode: 1968JAChS..90...99O.
- ↑ Douglas; McDaniel; Alexander (1994). Concepts and Models of Inorganic Chemistry (3rd ed.). John Wiley & Sons, Inc.. ISBN 978-0-471-62978-8.
- ↑ Puddephatt, R.A.; Stalteri, M.A. (1980). "Competition between Insertion of Sulfur Dioxide into the Methyl- or Phenyl- Transition Metal Bond". Journal of Organometallic Chemistry 193: C27–C29. doi:10.1016/S0022-328X(00)86091-X.
- ↑ Burger, B. J.; Thompson, M. E.; Cotter, W. D.; Bercaw, J. E. (1990). "Ethylene Insertion and β-hydrogen Elimination for Permethylscandocene Alkyl Complexes. A Study of the Chain Propagation and Termination Steps in Ziegler-Natta Polymerization of Ethylene". J. Am. Chem. Soc. 112 (4): 1566–1577. doi:10.1021/ja00160a041. Bibcode: 1990JAChS.112.1566B.
- ↑ Crabtree, R. H. (2009). The Organometallic Chemistry of the Transition Metals. John Wiley and Sons. p. 192. ISBN 978-0-470-25762-3.
- ↑ 15.0 15.1 15.2 Jones, Jane H. (2000). "The Cativa Process for the Manufacture of Acetic Acid". Platinum Metals Review 44 (3): 94–105. doi:10.1595/003214000X44394105. https://technology.matthey.com/article/44/3/94-105/.
- ↑ Sunley, G. J.; Watson, D. J. (2000). "High Productivity Methanol Carbonylation Catalysis using Iridium - The Cativa Process for the Manufacture of Acetic Acid". Catalysis Today 58 (4): 293–307. doi:10.1016/S0920-5861(00)00263-7.
- ↑ Kissin, Y. V. (2008). "Synthesis, Chemical Composition, and Structure of Transition Metal Components and Cocatalysts in Catalyst Systems for Alkene Polymerization". Alkene Polymerization Reactions with Transition Metal Catalysts. Amsterdam: Elsevier. pp. 207–290. ISBN 978-0-444-53215-2. https://books.google.com/books?id=JjhZiJEY5FsC&q=Alkene+Polymerization+Reactions+with+Transition+Metal+Catalysts&pg=PA207.
External links
 |  |
