Chemistry:Molecular orbital diagram
A molecular orbital diagram, or MO diagram, is a qualitative descriptive tool explaining chemical bonding in molecules in terms of molecular orbital theory in general and the linear combination of atomic orbitals (LCAO) method in particular.[1][2][3] A fundamental principle of these theories is that as atoms bond to form molecules, a certain number of atomic orbitals combine to form the same number of molecular orbitals, although the electrons involved may be redistributed among the orbitals. This tool is very well suited for simple diatomic molecules such as dihydrogen, dioxygen, and carbon monoxide but becomes more complex when discussing even comparatively simple polyatomic molecules, such as methane. MO diagrams can explain why some molecules exist and others do not. They can also predict bond strength, as well as the electronic transitions that can take place.
History
Qualitative MO theory was introduced in 1928 by Robert S. Mulliken[4][5] and Friedrich Hund.[6] A mathematical description was provided by contributions from Douglas Hartree in 1928[7] and Vladimir Fock in 1930.[8]
Basics
Molecular orbital diagrams are diagrams of molecular orbital (MO) energy levels, shown as short horizontal lines in the center, flanked by constituent atomic orbital (AO) energy levels for comparison, with the energy levels increasing from the bottom to the top. Lines, often dashed diagonal lines, connect MO levels with their constituent AO levels. Degenerate energy levels are commonly shown side by side. Appropriate AO and MO levels are filled with electrons by the Pauli Exclusion Principle, symbolized by small vertical arrows whose directions indicate the electron spins. The AO or MO shapes themselves are often not shown on these diagrams. For a diatomic molecule, an MO diagram effectively shows the energetics of the bond between the two atoms, whose AO unbonded energies are shown on the sides. For simple polyatomic molecules with a "central atom" such as methane (CH4) or carbon dioxide (CO2), a MO diagram may show one of the identical bonds to the central atom. For other polyatomic molecules, an MO diagram may show one or more bonds of interest in the molecules, leaving others out for simplicity. Often even for simple molecules, AO and MO levels of inner orbitals and their electrons may be omitted from a diagram for simplicity.
In MO theory molecular orbitals form by the overlap of atomic orbitals. Because σ bonds feature greater overlap than π bonds, σ bonding and σ* antibonding orbitals feature greater energy splitting (separation) than π and π* orbitals. The atomic orbital energy correlates with electronegativity as more electronegative atoms hold their electrons more tightly, lowering their energies. Sharing of molecular orbitals between atoms is more important when the atomic orbitals have comparable energy; when the energies differ greatly the orbitals tend to be localized on one atom and the mode of bonding becomes ionic. A second condition for overlapping atomic orbitals is that they have the same symmetry.
 |
| MO diagram for dihydrogen. Here electrons are shown by dots. |
|---|
Two atomic orbitals can overlap in two ways depending on their phase relationship (or relative signs for real orbitals). The phase (or sign) of an orbital is a direct consequence of the wave-like properties of electrons. In graphical representations of orbitals, orbital phase is depicted either by a plus or minus sign (which has no relationship to electric charge) or by shading one lobe. The sign of the phase itself does not have physical meaning except when mixing orbitals to form molecular orbitals.
Two same-sign orbitals have a constructive overlap forming a molecular orbital with the bulk of the electron density located between the two nuclei. This MO is called the bonding orbital and its energy is lower than that of the original atomic orbitals. A bond involving molecular orbitals which are symmetric with respect to any rotation around the bond axis is called a sigma bond (σ-bond). If the phase cycles once while rotating round the axis, the bond is a pi bond (π-bond). Symmetry labels are further defined by whether the orbital maintains its original character after an inversion about its center; if it does, it is defined gerade, g. If the orbital does not maintain its original character, it is ungerade, u.
Atomic orbitals can also interact with each other out-of-phase which leads to destructive cancellation and no electron density between the two nuclei at the so-called nodal plane depicted as a perpendicular dashed line. In this anti-bonding MO with energy much higher than the original AO's, any electrons present are located in lobes pointing away from the central internuclear axis. For a corresponding σ-bonding orbital, such an orbital would be symmetrical but differentiated from it by an asterisk as in σ*. For a π-bond, corresponding bonding and antibonding orbitals would not have such symmetry around the bond axis and be designated π and π*, respectively.
The next step in constructing an MO diagram is filling the newly formed molecular orbitals with electrons. Three general rules apply:
- The Aufbau principle states that orbitals are filled starting with the lowest energy
- The Pauli exclusion principle states that the maximum number of electrons occupying an orbital is two, with opposite spins
- Hund's rule states that when there are several MO's with equal energy, the electrons occupy the MO's one at a time before two electrons occupy the same MO.
The filled MO highest in energy is called the highest occupied molecular orbital (HOMO) and the empty MO just above it is then the lowest unoccupied molecular orbital (LUMO). The electrons in the bonding MO's are called bonding electrons and any electrons in the antibonding orbital would be called antibonding electrons. The reduction in energy of these electrons is the driving force for chemical bond formation. Whenever mixing for an atomic orbital is not possible for reasons of symmetry or energy, a non-bonding MO is created, which is often quite similar to and has energy level equal or close to its constituent AO, thus not contributing to bonding energetics. The resulting electron configuration can be described in terms of bond type, parity and occupancy for example dihydrogen 1σg2. Alternatively it can be written as a molecular term symbol e.g. 1Σg+ for dihydrogen. Sometimes, the letter n is used to designate a non-bonding orbital.
For a stable bond, the bond order defined as
must be positive.
The relative order in MO energies and occupancy corresponds with electronic transitions found in photoelectron spectroscopy (PES). In this way it is possible to experimentally verify MO theory. In general, sharp PES transitions indicate nonbonding electrons and broad bands are indicative of bonding and antibonding delocalized electrons. Bands can resolve into fine structure with spacings corresponding to vibrational modes of the molecular cation (see Franck–Condon principle). PES energies are different from ionisation energies which relates to the energy required to strip off the nth electron after the first n − 1 electrons have been removed. MO diagrams with energy values can be obtained mathematically using the Hartree–Fock method. The starting point for any MO diagram is a predefined molecular geometry for the molecule in question. An exact relationship between geometry and orbital energies is given in Walsh diagrams.
s-p mixing
The phenomenon of s-p mixing occurs when molecular orbitals of the same symmetry formed from the combination of 2s and 2p atomic orbitals are close enough in energy to further interact, which can lead to a change in the expected order of orbital energies.[9] When molecular orbitals are formed, they are mathematically obtained from linear combinations of the starting atomic orbitals. Generally, in order to predict their relative energies, it is sufficient to consider only one atomic orbital from each atom to form a pair of molecular orbitals, as the contributions from the others are negligible. For instance, in dioxygen the 3σg MO can be roughly considered to be formed from interaction of oxygen 2pz AOs only. It is found to be lower in energy than the 1πu MO, both experimentally and from more sophisticated computational models, so that the expected order of filling is the 3σg before the 1πu.[10] Hence the approximation to ignore the effects of further interactions is valid. However, experimental and computational results for homonuclear diatomics from Li2 to N2 and certain heteronuclear combinations such as CO and NO show that the 3σg MO is higher in energy than (and therefore filled after) the 1πu MO.[11] This can be rationalised as the first-approximation 3σg has a suitable symmetry to interact with the 2σg bonding MO formed from the 2s AOs. As a result, the 2σg is lowered in energy, whilst the 3σg is raised. For the aforementioned molecules this results in the 3σg being higher in energy than the 1πu MO, which is where s-p mixing is most evident. Likewise, interaction between the 2σu* and 3σu* MOs leads to a lowering in energy of the former and a raising in energy of the latter.[9] However this is of less significance than the interaction of the bonding MOs.
Diatomic MO diagrams
A diatomic molecular orbital diagram is used to understand the bonding of a diatomic molecule. MO diagrams can be used to deduce magnetic properties of a molecule and how they change with ionization. They also give insight to the bond order of the molecule, how many bonds are shared between the two atoms.[12]
The energies of the electrons are further understood by applying the Schrödinger equation to a molecule. Quantum Mechanics is able to describe the energies exactly for single electron systems but can be approximated precisely for multiple electron systems using the Born-Oppenheimer Approximation, such that the nuclei are assumed stationary. The LCAO-MO method is used in conjunction to further describe the state of the molecule. [13]
Diatomic molecules consist of a bond between only two atoms. They can be broken into two categories: homonuclear and heteronuclear. A homonuclear diatomic molecule is one composed of two atoms of the same element. Examples are H2, O2, and N2. A heteronuclear diatomic molecule is composed of two atoms of two different elements. Examples include CO, HCl, and NO.
Dihydrogen


The smallest molecule, hydrogen gas exists as dihydrogen (H-H) with a single covalent bond between two hydrogen atoms. As each hydrogen atom has a single 1s atomic orbital for its electron, the bond forms by overlap of these two atomic orbitals. In the figure the two atomic orbitals are depicted on the left and on the right. The vertical axis always represents the orbital energies. Each atomic orbital is singly occupied with an up or down arrow representing an electron.
Application of MO theory for dihydrogen results in having both electrons in the bonding MO with electron configuration 1σg2. The bond order for dihydrogen is (2-0)/2 = 1. The photoelectron spectrum of dihydrogen shows a single set of multiplets between 16 and 18 eV (electron volts).[14]
The dihydrogen MO diagram helps explain how a bond breaks. When applying energy to dihydrogen, a molecular electronic transition takes place when one electron in the bonding MO is promoted to the antibonding MO. The result is that there is no longer a net gain in energy.
The superposition of the two 1s atomic orbitals leads to the formation of the σ and σ* molecular orbitals. Two atomic orbitals in phase create a larger electron density, which leads to the σ orbital. If the two 1s orbitals are not in phase, a node between them causes a jump in energy, the σ* orbital. From the diagram you can deduce the bond order, how many bonds are formed between the two atoms. For this molecule it is equal to one. Bond order can also give insight to how close or stretched a bond has become if a molecule is ionized.[12]
Dihelium and diberyllium
Dihelium (He-He) is a hypothetical molecule and MO theory helps to explain why dihelium does not exist in nature. The MO diagram for dihelium looks very similar to that of dihydrogen, but each helium has two electrons in its 1s atomic orbital rather than one for hydrogen, so there are now four electrons to place in the newly formed molecular orbitals.

The only way to accomplish this is by occupying both the bonding and antibonding orbitals with two electrons, which reduces the bond order ((2−2)/2) to zero and cancels the net energy stabilization. However, by removing one electron from dihelium, the stable gas-phase species He+2 ion is formed with bond order 1/2.
Another molecule that is precluded based on this principle is diberyllium. Beryllium has an electron configuration 1s22s2, so there are again two electrons in the valence level. However, the 2s can mix with the 2p orbitals in diberyllium, whereas there are no p orbitals in the valence level of hydrogen or helium. This mixing makes the antibonding 1σu orbital slightly less antibonding than the bonding 1σg orbital is bonding, with a net effect that the whole configuration has a slight bonding nature. This explains the fact that the diberyllium molecule exists and has been observed in the gas phase.[15][16] The slight bonding nature explains the low dissociation energy of only 59 kJ·mol−1.[15]
Dilithium
MO theory correctly predicts that dilithium is a stable molecule with bond order 1 (configuration 1σg21σu22σg2). The 1s MOs are completely filled and do not participate in bonding.

Dilithium is a gas-phase molecule with a much lower bond strength than dihydrogen because the 2s electrons are further removed from the nucleus. In a more detailed analysis[17] which considers the environment of each orbital due to all other electrons, both the 1σ orbitals have higher energies than the 1s AO and the occupied 2σ is also higher in energy than the 2s AO (see table 1).
Diboron
The MO diagram for diboron (B-B, electron configuration 1σg21σu22σg22σu21πu2) requires the introduction of an atomic orbital overlap model for p orbitals. The three dumbbell-shaped p-orbitals have equal energy and are oriented mutually perpendicularly (or orthogonally). The p-orbitals oriented in the z-direction (pz) can overlap end-on forming a bonding (symmetrical) σ orbital and an antibonding σ* molecular orbital. In contrast to the sigma 1s MO's, the σ 2p has some non-bonding electron density at either side of the nuclei and the σ* 2p has some electron density between the nuclei.
The other two p-orbitals, py and px, can overlap side-on. The resulting bonding orbital has its electron density in the shape of two lobes above and below the plane of the molecule. The orbital is not symmetric around the molecular axis and is therefore a pi orbital. The antibonding pi orbital (also asymmetrical) has four lobes pointing away from the nuclei. Both py and px orbitals form a pair of pi orbitals equal in energy (degenerate) and can have higher or lower energies than that of the sigma orbital.
In diboron the 1s and 2s electrons do not participate in bonding but the single electrons in the 2p orbitals occupy the 2πpy and the 2πpx MO's resulting in bond order 1. Because the electrons have equal energy (they are degenerate) diboron is a diradical and since the spins are parallel the molecule is paramagnetic.

In certain diborynes the boron atoms are excited and the bond order is 3.
Dicarbon
Like diboron, dicarbon (C-C electron configuration:1σg21σu22σg22σu21πu4) is a reactive gas-phase molecule. The molecule can be described as having two pi bonds but without a sigma bond.[18]
Dinitrogen

With nitrogen, we see the two molecular orbitals mixing and the energy repulsion. This is the reasoning for the rearrangement from a more familiar diagram. Notice how the σ from the 2p behaves more non-bonding like due to mixing, same with the 2s σ. This also causes a large jump in energy in the 2p σ* orbital. The bond order of diatomic nitrogen is three, and it is a diamagnetic molecule.[12]
The bond order for dinitrogen (1σg21σu22σg22σu21πu43σg2) is three because two electrons are now also added in the 3σ MO. The MO diagram correlates with the experimental photoelectron spectrum for nitrogen.[19] The 1σ electrons can be matched to a peak at 410 eV (broad), the 2σg electrons at 37 eV (broad), the 2σu electrons at 19 eV (doublet), the 1πu4 electrons at 17 eV (multiplets), and finally the 3σg2 at 15.5 eV (sharp).
Dioxygen

Oxygen has a similar setup to H2, but now we consider 2s and 2p orbitals. When creating the molecular orbitals from the p orbitals, notice the three atomic orbitals split into three molecular orbitals, a singly degenerate σ and a doubly degenerate π orbital. Another property we can observe by examining molecular orbital diagrams is the magnetic property of diamagnetic or paramagnetic. If all the electrons are paired, there is a slight repulsion and it is classified as diamagnetic. If unpaired electrons are present, it is attracted to a magnetic field, and therefore paramagnetic. Oxygen is an example of a paramagnetic diatomic. Also notice the bond order of diatomic oxygen is two. [12]
MO treatment of dioxygen is different from that of the previous diatomic molecules because the pσ MO is now lower in energy than the 2π orbitals. This is attributed to interaction between the 2s MO and the 2pz MO.[20] Distributing 8 electrons over 6 molecular orbitals leaves the final two electrons as a degenerate pair in the 2pπ* antibonding orbitals resulting in a bond order of 2. As in diboron, these two unpaired electrons have the same spin in the ground state, which is a paramagnetic diradical triplet oxygen. The first excited state has both HOMO electrons paired in one orbital with opposite spins, and is known as singlet oxygen.

The bond order decreases and the bond length increases in the order O+2 (112.2 pm), O2 (121 pm), O−2 (128 pm) and O2−2 (149 pm).[20]
Difluorine and dineon

In difluorine two additional electrons occupy the 2pπ* with a bond order of 1. In dineon Ne2 (as with dihelium) the number of bonding electrons equals the number of antibonding electrons and this molecule does not exist.
Dimolybdenum and ditungsten

Dimolybdenum (Mo2) is notable for having a sextuple bond. This involves two sigma bonds (4dz2 and 5s), two pi bonds (using 4dxz and 4dyz), and two delta bonds (4dx2 − y2 and 4dxy). Ditungsten (W2) has a similar structure.[21][22]
MO energies overview
Table 1 gives an overview of MO energies for first row diatomic molecules calculated by the Hartree-Fock-Roothaan method, together with atomic orbital energies.
| Table 1. Calculated MO energies for diatomic molecules in Hartrees [17] | |||||||
|---|---|---|---|---|---|---|---|
| H2 | Li2 | B2 | C2 | N2 | O2 | F2 | |
| 1σg | -0.5969 | -2.4523 | -7.7040 | - 11.3598 | - 15.6820 | - 20.7296 | -26.4289 |
| 1σu | -2.4520 | -7.7032 | -11.3575 | -15.6783 | -20.7286 | -26.4286 | |
| 2σg | -0.1816 | -0.7057 | -1.0613 | -1.4736 | -1.6488 | -1.7620 | |
| 2σu | -0.3637 | -0.5172 | -0.7780 | -1.0987 | -1.4997 | ||
| 3σg | -0.6350 | -0.7358 | -0.7504 | ||||
| 1πu | -0.3594 | -0.4579 | -0.6154 | -0.7052 | -0.8097 | ||
| 1πg | -0.5319 | -0.6682 | |||||
| 1s (AO) | -0.5 | -2.4778 | -7.6953 | -11.3255 | -15.6289 | -20.6686 | -26.3829 |
| 2s (AO) | -0.1963 | -0.4947 | -0.7056 | -0.9452 | -1.2443 | -1.5726 | |
| 2p (AO) | -0.3099 | -0.4333 | -0.5677 | -0.6319 | -0.7300 |
Heteronuclear diatomics
In heteronuclear diatomic molecules, mixing of atomic orbitals only occurs when the electronegativity values are similar. In carbon monoxide (CO, isoelectronic with dinitrogen) the oxygen 2s orbital is much lower in energy than the carbon 2s orbital and therefore the degree of mixing is low. The electron configuration 1σ21σ*22σ22σ*21π43σ2 is identical to that of nitrogen. The g and u subscripts no longer apply because the molecule lacks a center of symmetry.
In hydrogen fluoride (HF), the hydrogen 1s orbital can mix with fluorine 2pz orbital to form a sigma bond because experimentally the energy of 1s of hydrogen is comparable with 2p of fluorine. The HF electron configuration 1σ22σ23σ21π4 reflects that the other electrons remain in three lone pairs and that the bond order is 1.
The more electronegative atom is the more energetically excited because it more similar in energy to its atomic orbital. This also accounts for the majority of the electron negativity residing around the more electronegative molecule. Applying the LCAO-MO method allows us to move away from a more static Lewis structure type approach and actually account for periodic trends that influence electron movement. Non-bonding orbitals refer to lone pairs seen on certain atoms in a molecule. A further understanding for the energy level refinement can be acquired by delving into quantum chemistry; the Schrödinger equation can be applied to predict movement and describe the state of the electrons in a molecule.[13][23]
NO

Nitric oxide is a heteronuclear molecule that exhibits mixing. The construction of its MO diagram is the same as for the homonuclear molecules. It has a bond order of 2.5 and is a paramagnetic molecule. The energy differences of the 2s orbitals are different enough that each produces its own non-bonding σ orbitals. Notice this is a good example of making the ionized NO+ stabilize the bond and generate a triple bond, also changing the magnetic property to diamagnetic.[12]
HF

Hydrogen fluoride is another example of a heteronuclear molecule. It is slightly different in that the π orbital is non-bonding, as well as the 2s σ. From the hydrogen, its valence 1s electron interacts with the 2p electrons of fluorine. This molecule is diamagnetic and has a bond order of one.
Triatomic molecules
Carbon dioxide
Carbon dioxide, CO2, is a linear molecule with a total of sixteen bonding electrons in its valence shell. Carbon is the central atom of the molecule and a principal axis, the z-axis, is visualized as a single axis that goes through the center of carbon and the two oxygens atoms. For convention, blue atomic orbital lobes are positive phases, red atomic orbitals are negative phases, with respect to the wave function from the solution of the Schrödinger equation.[24] In carbon dioxide the carbon 2s (−19.4 eV), carbon 2p (−10.7 eV), and oxygen 2p (−15.9 eV)) energies associated with the atomic orbitals are in proximity whereas the oxygen 2s energy (−32.4 eV) is different.[25]
Carbon and each oxygen atom will have a 2s atomic orbital and a 2p atomic orbital, where the p orbital is divided into px, py, and pz. With these derived atomic orbitals, symmetry labels are deduced with respect to rotation about the principal axis which generates a phase change, pi bond (π)[26] or generates no phase change, known as a sigma bond (σ).[27] Symmetry labels are further defined by whether the atomic orbital maintains its original character after an inversion about its center atom; if the atomic orbital does retain its original character it is defined gerade, g, or if the atomic orbital does not maintain its original character, ungerade, u. The final symmetry-labeled atomic orbital is now known as an irreducible representation.
Carbon dioxide’s molecular orbitals are made by the linear combination of atomic orbitals of the same irreducible representation that are also similar in atomic orbital energy. Significant atomic orbital overlap explains why sp bonding may occur.[28] Strong mixing of the oxygen 2s atomic orbital is not to be expected and are non-bonding degenerate molecular orbitals. The combination of similar atomic orbital/wave functions and the combinations of atomic orbital/wave function inverses create particular energies associated with the nonbonding (no change), bonding (lower than either parent orbital energy) and antibonding (higher energy than either parent atomic orbital energy) molecular orbitals.
- MO model carbon dioxide
-
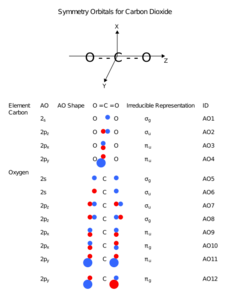 Atomic orbitals of carbon dioxide
Atomic orbitals of carbon dioxide -
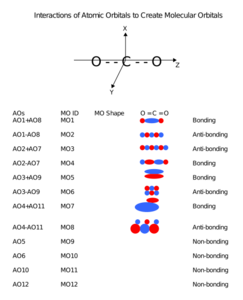 Molecular orbitals of carbon dioxide
Molecular orbitals of carbon dioxide -
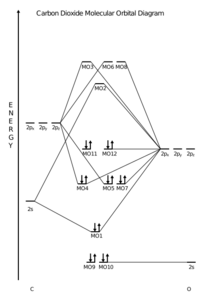 MO diagram of carbon dioxide
MO diagram of carbon dioxide



Water
For nonlinear molecules, the orbital symmetries are not σ or π but depend on the symmetry of each molecule. Water (H2O) is a bent molecule (105°) with C2v molecular symmetry. The possible orbital symmetries are listed in the table below. For example, an orbital of B1 symmetry (called a b1 orbital with a small b since it is a one-electron function) is multiplied by -1 under the symmetry operations C2 (rotation about the 2-fold rotation axis) and σv'(yz) (reflection in the molecular plane). It is multiplied by +1(unchanged) by the identity operation E and by σv(xz) (reflection in the plane bisecting the H-O-H angle).

| C2v | E | C2 | σv(xz) | σv'(yz) | ||
|---|---|---|---|---|---|---|
| A1 | 1 | 1 | 1 | 1 | z | x2, y2, z2 |
| A2 | 1 | 1 | −1 | −1 | Rz | xy |
| B1 | 1 | −1 | 1 | −1 | x, Ry | xz |
| B2 | 1 | −1 | −1 | 1 | y, Rx | yz |
The oxygen atomic orbitals are labeled according to their symmetry as a1 for the 2s orbital and b1 (2px), b2 (2py) and a1 (2pz) for the three 2p orbitals. The two hydrogen 1s orbitals are premixed to form a1 (σ) and b2 (σ*) MO.
Mixing takes place between same-symmetry orbitals of comparable energy resulting a new set of MO's for water:
- 2a1 MO from mixing of the oxygen 2s AO and the hydrogen σ MO.
- 1b2 MO from mixing of the oxygen 2py AO and the hydrogen σ* MO.
- 3a1 MO from mixing of the a1 AOs.
- 1b1 nonbonding MO from the oxygen 2px AO (the p-orbital perpendicular to the molecular plane).
In agreement with this description the photoelectron spectrum for water shows a sharp peak for the nonbonding 1b1 MO (12.6 eV) and three broad peaks for the 3a1 MO (14.7 eV), 1b2 MO (18.5 eV) and the 2a1 MO (32.2 eV).[29] The 1b1 MO is a lone pair, while the 3a1, 1b2 and 2a1 MO's can be localized to give two O−H bonds and an in-plane lone pair.[30] This MO treatment of water does not have two equivalent rabbit ear lone pairs.[31]
Hydrogen sulfide (H2S) too has a C2v symmetry with 8 valence electrons but the bending angle is only 92°. As reflected in its photoelectron spectrum as compared to water the 5a1 MO (corresponding to the 3a1 MO in water) is stabilised (improved overlap) and the 2b2 MO (corresponding to the 1b2 MO in water) is destabilized (poorer overlap).
References
- ↑ Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2001). Organic Chemistry (1st ed.). Oxford University Press. pp. 96–103. ISBN 978-0-19-850346-0.
- ↑ Organic Chemistry, Third Edition, Marye Anne Fox, James K. Whitesell, 2003, ISBN:978-0-7637-3586-9
- ↑ Organic Chemistry 3rd Ed. 2001, Paula Yurkanis Bruice, ISBN:0-13-017858-6
- ↑ Mulliken, R. (1928). "The Assignment of Quantum Numbers for Electrons in Molecules. I". Physical Review 32 (2): 186–222. doi:10.1103/PhysRev.32.186. Bibcode: 1928PhRv...32..186M.
- ↑ Mulliken, R. (1928). "Electronic States and Band Spectrum Structure in Diatomic Molecules. VII. P2→S2 and S2→P2 Transitions". Physical Review 32 (3): 388–416. doi:10.1103/PhysRev.32.388. Bibcode: 1928PhRv...32..388M.
- ↑ Hund, F. (1928). "Zur Deutung der Molekelspektren. IV" (in de). Zeitschrift für Physik (Springer Science and Business Media LLC) 51 (11–12): 759–795. doi:10.1007/bf01400239. ISSN 1434-6001. Bibcode: 1928ZPhy...51..759H.
- ↑ Hartree, D. R. (1928). "The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part I. Theory and Methods". Mathematical Proceedings of the Cambridge Philosophical Society (Cambridge University Press (CUP)) 24 (1): 89–110. doi:10.1017/s0305004100011919. ISSN 0305-0041. Bibcode: 1928PCPS...24...89H. http://elib.bsu.by/handle/123456789/154382.
- ↑ Fock, V. (1930). "Näherungsmethode zur Lösung des quantenmechanischen Mehrkörperproblems" (in de). Zeitschrift für Physik (Springer Science and Business Media LLC) 61 (1–2): 126–148. doi:10.1007/bf01340294. ISSN 1434-6001. Bibcode: 1930ZPhy...61..126F.
- ↑ 9.0 9.1 Keeler, James; Wothers, Peter (2014). Chemical Structure and Reactivity - an integrated approach (2nd ed.). Oxford University Press. pp. 123–126. ISBN 978-0-19-9604135.
- ↑ Douglas, Bodie; McDaniel, Darl; Alexander, John (1994). Concepts and Models of Inorganic Chemistry (3rd ed.). Wiley. pp. 157–159. ISBN 978-0-471-62978-8.
- ↑ Sethi, M.S.; Satake, M. (1999). Chemical Bonding. New Delhi: Discovery Publishing House. pp. 93–95. ISBN 81-7141-163-0.
- ↑ 12.0 12.1 12.2 12.3 12.4 Pfennig, Brian (2015). Principles of Inorganic Chemistry. Hoboken, New Jersey: John Wiley & Sons, Inc.. ISBN 9781118859100.
- ↑ 13.0 13.1 McQuarrie, Donald A. (2008). Quantum chemistry (2nd ed.). Sausalito, Calif.: University Science Books. ISBN 9781891389504.
- ↑ .hydrogen @ PES database arizona.edu
- ↑ 15.0 15.1 Keeler, James; Wothers, Peter (2003). Why Chemical Reactions Happen. Oxford University Press. p. 74. ISBN 9780199249732.
- ↑ Merritt, Jeremy M.; Bondybey, Vladimir E.; Heaven, Michael C. (2009). "Beryllium Dimer—Caught in the Act of Bonding". Science 324 (5934): 1548–1551. doi:10.1126/science.1174326. PMID 19460963. Bibcode: 2009Sci...324.1548M. https://www.science.org/doi/10.1126/science.1174326.
- ↑ 17.0 17.1 Harrison, J. F.; Lawson, D. B. (2005). "Some Observations on Molecular Orbital Theory". Journal of Chemical Education 82 (8): 1205. doi:10.1021/ed082p1205. Bibcode: 2005JChEd..82.1205L.
- ↑ Shaik, S.; Rzepa, H. S.; Hoffmann, R. (2013). "One Molecule, Two Atoms, Three Views, Four Bonds?". Angew. Chem. Int. Ed. 52 (10): 3020–3033. doi:10.1002/anie.201208206. PMID 23362052.
- ↑ Bock, H.; Mollere, P. D. (1974). "Photoelectron spectra. An experimental approach to teaching molecular orbital models". Journal of Chemical Education 51 (8): 506. doi:10.1021/ed051p506. Bibcode: 1974JChEd..51..506B.
- ↑ 20.0 20.1 Modern Inorganic Chemistry William L. Jolly (McGraw-Hill 1984), p.106 ISBN:0-07-032760-2
- ↑ Roos, Björn O.; Borin, Antonio C.; Gagliardi, Laura (2007). "Reaching the Maximum Multiplicity of the Covalent Chemical Bond". Angewandte Chemie International Edition 46 (9): 1469–1472. doi:10.1002/anie.200603600. PMID 17225237. https://archive-ouverte.unige.ch/unige:3199/ATTACHMENT01.
- ↑ Frenking, Gernot; Tonner, Ralf (2007). "The six-bond bound". Nature 446 (7133): 276–277. doi:10.1038/446276a. PMID 17361173.
- ↑ Miessler, Gary (2014). Inorganic chemistry (Fifth ed.). Upper Saddle River, New Jersey: Pearson. ISBN 9781269453219.
- ↑ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. p. 9. ISBN 978-0-13-175553-6.
- ↑ "An Introduction to Molecular Orbitals". Jean & volatron. ""1993"" ISBN:0-19-506918-8. p.192
- ↑ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. p. 38. ISBN 978-0-13-175553-6.
- ↑ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. p. 34. ISBN 978-0-13-175553-6.
- ↑ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. p. 33. ISBN 978-0-13-175553-6.
- ↑ Levine, I. N. (1991). Quantum Chemistry (4th ed.). Prentice-Hall. p. 475. ISBN 0-7923-1421-2.
- ↑ Jochen Autschbach (2012). "Orbitals: Some Fiction and Some Facts". Journal of Chemical Education 89 (8): 1032–1040. doi:10.1021/ed200673w. Bibcode: 2012JChEd..89.1032A.
- ↑ Laing, Michael (1987). "No rabbit ears on water. The structure of the water molecule: What should we tell the students?". Journal of Chemical Education 64 (2): 124. doi:10.1021/ed064p124. Bibcode: 1987JChEd..64..124L.
External links
- MO diagrams at meta-synthesis.com Link
- MO diagrams at chem1.com Link
- Molecular orbitals at winter.group.shef.ac.uk Link
 |  |


{kind=link}