Medicine:Liposarcoma
| Liposarcoma | |
|---|---|
 | |
| Histopathology of liposarcoma, H&E stain:[1] - | |
| Specialty | Dermatology, general surgery oncology |
| Symptoms | Lump under skin, pain, swelling, organ dysfunction |
Liposarcomas are the most common subtype of soft tissue sarcomas, accounting for at least 20% of all sarcomas in adults.[2] Soft tissue sarcomas are rare neoplasms with over 150 different histological subtypes or forms. Liposarcomas arise from the precursor lipoblasts of the adipocytes (i.e. fat cells) in adipose (i.e. fat) tissues. Adipose tissues are distributed throughout the body, including such sites as the deep and more superficial layers of subcutaneous tissues as well as in less surgically accessible sites like the retroperitoneum (i.e. space behind the abdominal cavity) and visceral fat inside the abdominal cavity.[3]
All liposarcomas consist of at least some cells that bear a resemblance to fat cells when examined for their histopathologic appearances under a microscope.[4] However, the liposarcomas do have several forms based on differences in their clinical presentations (e.g. ages, gender preferences, sites of tumors, signs, and symptoms), severities (i.e. potential to invade local tissues, recur after surgical removal, and metastasize to distal tissues), genetic abnormalities, prognoses, and preferred treatment regimens. The World Health Organization in 2020 reclassified liposarcomas into five more or less distinct forms: 1) atypical lipomatous tumor/well-differentiated liposarcoma (WD-LPS); 2) dedifferentiated liposarcoma (DD-LPS); 3) myxoid liposarcoma; 4) pleomorphic liposarcoma; and 5) myxoid pleomorphic liposarcoma.[5] (Pleomorphic indicates the presence of cells that have abnormal and often large variations in their size and shape and/or the size and shape of their nuclei.)
While liposarcoma forms are classified as being aggressive and malignant or, in the case of the atypical lipomatous tumor/well-differentiated liposarcoma, as relatively non-aggressive and benign,[6] all five liposarcoma forms can infiltrate locally to injure nearby tissues and organs, occur in surgically inaccessible sites adjacent to vital organs (e.g. the retroperitoneum[7]), recur after surgical removal, and progress to life-threatening diseases. Studies to date find that all five liposarcoma forms, while usually treatable at least initially by surgical resection, are often only marginally responsive to currently used chemotherapy and radiotherapy regimens. The liposarcomas require a wide range of further studies to determine their responsiveness to various radiotherapy, chemotherapy, and more novel treatment regimens as used individually and in various combinations that would include, where possible, surgical removal.[6]
Etymology
"fatty tumor" (plural lipomata), 1830, medical Latin, from Greek lipos "fat" (n.), from PIE root *leip- "to stick, adhere", also used to form words for "fat", + -oma.
1650s, "fleshy excrescence", (plural liposarcomata), Medical Latin, from Latinized form of Greek sarkoma "fleshy substance" (Galen), from sarkoun "to produce flesh, grow fleshy", from sarx (genitive sarkos) "flesh", + -oma.
Forms of liposarcomas

Liposarcomas are generally large tumors (>10 cm) but can be of almost any size. They occur mainly in adults with only 0.7% of cases occurring in those <16 years old.[5] In adults, liposarcomas occur predominantly in and after middle-age.[8] The very rare cases occurring in children and adolescents are diagnosed predominantly as being the myxoid liposarcoma form.[5]
The five liposarcoma forms must be distinguished not only from each other but also from certain other soft tissue tumors. These other tumors along with some of their distinguishing histopathologic features are: 1) dysplastic lipomas (i.e. benign humors that have sites of tissue necrosis and neoplastic, variably-sized fat cells containing variable sized/shaped nuclei; these neoplastic cells, unlike most neoplastic cell in the liposarcomas, do not overexpress the MDM2 gene);[9] 2) atypical spindle cell lipomas (i.e. benign tumors with mildly atypical spindle-shaped cells in a fibrous-to-myxoid stroma intermixed with vacuolated lipoblasts and variable-sized adipocytes with atypical nuclei; 3) pleomorphic lipomas (i.e. benign tumors characterized by giant cells with overlapping nuclei);[8] and 4) solitary fibrous tumors (i.e. tumors, up to 22% of which exhibit malignant behavior, consisting of spindle- or ovoid-shape cells within a collagenous background stroma intermixed with blood vessels with a characteristic staghorn shape[10]).[5]
Atypical lipomatous tumor/well-differentiated liposarcoma
Together, atypical lipomatous tumors (ALTs) and well-differentiated liposarcomas (WDLs) account for 40–45% of all liposarcomas.[11] They rarely if ever metastasize and therefore are regarded as benign or premalignant tumors.[12][13] However, they are locally invasive and may transform to a more aggressive and potentially metastasizing liposarcoma, i.e. a dedifferentiated liposarcoma. Furthermore, a surgically removed atypical lipomatous tumor/well-differentiated liposarcoma may recur as a dedifferentiated liposarcoma.[6]
Presentation

ALTs and WDLs are considered virtually identical tumors except that by definition ALTs designate tumors that develop in the arms or legs while WDLs designate tumors that develop in less surgically accessible sites such as the deep, centrally-located soft tissues of the retroperitoneum, paratesticular region (i.e. area within the scrotum including the testes, spermatic cord, testicular tunic, epididymis, and appendix of testis),[6] oral cavity, and eye socket.[12][14] This terminology has prognostic implications: less than 7% of ALT tumors convert to dedifferentiated liposarcomas within a median time of 7 years while 17% of WDL tumors convert to this more malignant liposarcoma within a median time of 8 years.[6] ALT and WDL (hereafter termed ALT/WDL) tumors typically present in middle-aged and older individuals as slowly enlarging masses that tend to be larger and at a more advanced stage when located in deep tissues.[8][11] These tumors usually are not painful and if located superficially, readily apparent; they can also cause extensive edema (i.e. swelling due to the local accumulation of fluid) in involved areas such as the thigh (see adjacent figure) due to their invasion into the blood and/or lymphatic vessels draining the tumor's site. Deep-seated ALT/WDL tumors may be asymptomatic but, depending on their location, produce serious signs and/or symptoms of disfunction in any one of the various organs which they infiltrate. These organs include those close to or in the retroperitoneum (e.g. intestines, kidney, and the kidney's ureters); the paratesticular region; the mediastinum (e.g. trachea and lung's major bronchi); and the head (e.g. the retrobulbar space behind the globe of the eye).[12][14]
Pathology

Histopathologically, ALT/WDL tumors are divided into adipocytic/lipoma-like, sclerosing, and inflammatory variants with adipocyte/lipoma-like being the most common. Adipocytic/lipoma-like ALT/WDL tumors consist of lobules of mature fat cells variably intersected with irregular fibrous septa (see the adjacent H&E stained photomicrograph). Sclerosing ALT/WDL tumors, the second most common variant, develop primarily in the retroperitoneal and paratesticular areas; it consists of scattered, atypical stromal cells within a collagenous (i.e. collagen-containing) stromal tissue background. Rare vacuole-containing lipoblasts populate this tissue. Inflammatory ALT/WDL tumors are the rarest variant. they occur most frequently in the retroperitoneum and consists of chronic inflammatory cells, e.g. lymphocytes and plasma cells plus occasional lymph node-like follicles interspersed throughout a tissue background containing fat cells.[14]
Genetics
The neoplastic cells in ALT/WDL tumors contain one or more extra ring-shaped small supernumerary marker chromosome (sSMC) or an abnormal giant marker chromosome (i.e. a formerly normal chromosome that is made abnormal by having a duplication of parts of its own or one or more other chromosome's genetic material). These abnormal chromosomes contain extra copies of chromosome 12's long arm (also termed the q arm) at bands 13 through 15. This stretch of chromosome 12 includes the MDM2 proto-oncogene (a potentially tumor-causing gene when overexpressed) located at band 15[15] and CDK4 (a gene that when overexpressed promotes the development of various tumors) located at band 14.1.[16][17] The amplification (i.e. increased copies of a gene without a proportional increase in other genes) of these two genes is a highly sensitive and specific indicator that a liposarcoma is either an ALT/WDL or a dedifferentiated liposarcoma rather than any other liposarcoma or lipoma form.[17] In addition to the MDM2 and CDK4 genes, this band 13–15 chromosome area also contains the TSPAN31 and HMGA2 genes which, when overexpressed, are associated with various tumors and/or cancers. One or more of these overexpressed genes, it has been suggested, promote and/or contribute to the development and/or progression of ALT/WDL tumors.[8]
Diagnosis
The diagnosis of ALT/WDL tumors is made based on the features of their clinical presentations, histopathology, and genetic findings. In particular, detection in the ALT/WDL tumor cells of an overexpressed MDM2 or CDK4 gene or the presence of either the specific ALT/WDL-associated sSMC or giant marker chromosome (as defined by next generation DNA sequencing, comparative genomic hybridization,[18] and/or highly specialized cytogenetic G banding analyses[19]) strongly supports the diagnosis of ALT/WDL or dedifferentiated liposarcoma. The clinical presentation and histopathology differences between the latter two liposarcoma forms usually help distinguish between them.[8]
Treatment and prognosis
ALT/WDL tumors are treated by radical surgical resection to remove all tumor neoplastic tissues. However, these tumors recur locally in 30–50% of cases. Recurrences occur most often in tumors located in less accessible sites such those in the retroperitoneum, mediastinum, and spermatic cord. These less surgically assessible tumors tend to recur repeatedly and ultimately may cause death due to their injurious effects on vital organs. While ALT/WDL tumors have very little potential to metastasize, about 10% will convert to an overtly malignant and potentially metastasizing liposarcoma form, dedifferentiated liposarcoma. The median time for this malignant transformation is about 7–9 years.[8] In addition, a surgically removed ALT/WDL may recur after a variable interval as a dedifferentiated liposarcoma.[6] A large randomized controlled trial comparing radiotherapy followed by surgery to surgery alone in ALT/WDL tumors found little difference between the two regimens. Smaller studies employing selective inhibitors of the protein products of the CDK4 or MDM2 genes implicated in ALT/WDL have shown at best only modest effects. Further studies using these or completely novel treatment regimens are under investigation.[8] A review study in 2012 reported the 5 and 10 year survival rates of individuals with ALT/WDL to be 100% and 87%, respectively.[20]
Novel therapies
The novel therapies of ALT/WDL are the same as those listed in the Novel therapies section of Dedifferentiated liposarcoma.[citation needed]
Dedifferentiated liposarcoma
Dedifferentiated liposarcomas are malignant tumors which in ~10% of cases develop in an existing atypical lipomatous tumor/well-differentiated liposarcoma (ALT/WDL) tumor or at the site were an ALT/WPL tumor was surgically removed. Individuals with a de novo diagnosis of this tumor may have had an ALT/WDL that progressed to a dedifferentiated liposarcoma but went undetected because it developed asymptomatically in a highly sequestered site such as the retroperitoneum or abdominal cavity. Many of the dedifferentiated liposarcoma tumors' clinical and genetic features are similar to those found in ALT/WDL tumors.[8]
Presentation
Dedifferentiated lipoosarcomas (DDL) occur most frequently in middle-aged and older adults with a peak incidence in their sixth to eighth decades.[8] Rarely, these tumors have developed in children and adolescents.[5] DDL tumors most commonly occur in the retroperitoneal space but, similar to ALT/WDL, may occur in the extremities, paratesticular area, mediastinum, head, or neck.[8] Less than 1% of all DDLs develop in superficial soft tissues[8] or the eye socket.[21] At presentation, DDL tumors typically are painless, large, may have been slowly and progressively enlarging for years,[8] and on routine X-rays contain areas of calcium deposition (exemplified by Fig. 1 in the Histopathology of liposarcomas section).[22][23] Less commonly, affected individuals have signs and/or symptoms due to their tumor's impingement on an organ (e.g. abdominal pain caused by blockage of the intestines or urinary tract obstruction caused by blockage of the urethra). Very rarely, individuals with DDL present with one or more signs or symptoms of chronic inflammation (see B symptoms) and/or one of the endrocrine, neurological, mucocutaneous, hematological, or other tissue-related paraneoplastic syndromes. The signs and symptoms of chronic inflammation and the various paraneoplastic syndromes are caused by the tumors' secretion of cytokines, hormones, prostaglandins, and/or other systemically acting agents; they completely disappear after the DDL is successfully treated.[8]
Pathology
The histopathological appearance of DDL tumors (see Fig. 2 in the below Histopathology of liposarcomas section) varies widely but most frequently exhibits features of undifferentiated pleomorphic sarcomas (which are tumors densely populated with variably sized and shaped cells containing variability sized and shaped nuclei) or spindle cell sarcomas (which are tumors consisting of spindle-shaped cells in a connective tissue background). Different parts of DDL tumors often show variations in the appearances of their background connective tissues: these tissues may be myxoid (i.e. consisting of a clear, mucus-like substance which when stained using a standard H&E stain method appears more blue or purple than the red color of normal tissues) or myxocollagenous (i.e. high collagen fiber content in a myxoid background), and, in ~5% of cases, have areas of osteoid (see Fig. 1 in the below Histopathology of liposarcomas section) or cartilaginous material. The tumors also show large variations in their cell contents. For example, up to 10% of DDL tumors have areas with ALT/WDL histopathology [8] and rare cases of DDL have areas containing meningothelial-like whorls of flat cells.[24][25]
Genetics
The neoplastic cells in both DDL and ALT/WDL carry similar small supernumerary marker chromosomes (sSMCs) and/or giant marker chromosomes that contain extra parts of chromosome 12's q arm at bands 13 through 15. This chromosomal area includes two genes associated with tumor development, the MDM2[26] and CDK4 genes.[27][17] The presence of extra copies of these two genes and/or their overproduced protein products is a highly sensitive and specific indicator that a lipomatous tumor is an ALT/WDL or DDL rather than some other type of lipomatous tumor.[12][17] Overexpression of the MDM2 and CDK genes, and/or other genetic material in the sSMCs or giant marker chromosomes are suspected of promoting the development and/or progression of DDL as well as ALT/WDL tumors.[28] Other genes in the sMMC and giant marker chromosome that are also overexpressed in ALT/WDL and DDL neoplastic cells include HMGA2, CPM, YEATS4,[29] and DDIT3. Compared to ALT/WDL neoplastic cells, however, DDL neoplastic cells: 1) express higher levels of the genes in the two abnormal chromosomes; this may contribute to the progression of ALT/WDL to DDL; and 2) higher levels of gene products on the long arm of chromosome 1 at band 32, the long arm of chromosome 6 at band 33, and, in ~25% of cases, the short arm of chromosome 1 at band 32.2 which contains the JUN gene (this gene is overexpressed in DDL but not ALT/WDL). Since the JUN gene's product, c-jun, inhibits cell death and promotes cell proliferation, its overproduction may contribute to the progression of ALT/WDL to DDL and/or the malignancy of DDL neoplastic cells.[8] Gene expression profiling (i.e. measurement of the expression of the products of thousands of genes made by cells, tissues, or tumors) have revealed that adipocyte cell differentiation and metabolic pathways in ALT/WDL are upregulated while cell proliferation and DNA damage response pathways are upregulated in DDL.[6]
Diagnosis
The histopathological of DDL is often insufficiently clear to make a firm diagnosis. However, the diagnosis of DDL is supported in individuals: whose tumors contain ALT/WDL admixed with DDL histological components; with histories of having a prior ALT/WDL;[8] or who present with a retroperitoneal liposarcoma (DDL constitutes ~57% of all retroperitoneal liposarcomas). DDL tumors only rarely (<1% of cases) present as superficial skin tumors;[8] are almost 5 times less likely than ALT/WDL to occur in the eye socket;[14][21] and are extremely rare in children.[5] Detection of tumor cell MDM2 amplification is the diagnostic gold standard in distinguishing WDL from lipomas, dysplastic lipomas, atypical spindle cell sarcomas, pleomorphic lipomas, and solitary fibrous tumors.[8] Alternately, detection in the tumor cells of an overexpressed CDK4 gene or the presence of either the specific ALT/WDL-associated sSMCs or giant marker chromosome strongly support the diagnosis of DDL or ALT/WDL.[18][19] The clinical presentation, histopathology, and gene differences (e.g. tumor cell overexpression of the cJUN gene strongly favors the diagnosis of DDL over ATL/WDL) between the latter two liposarcoma forms usually help distinguish between them.[8]
Treatment and Prognosis
Complete surgical resection is usually the recommended first-line treatment for localized DDL tumors.[6] However, emerging studies suggest that patients with DDL tumors that are restricted to an extremity or the trunk and have a predicted 10-year tumor-related overall survival of 51% or less have improved outcomes when chemotherapy (e.g. doxorubicin plus ifosfamide) is added to their surgical regimens.[30] For these localized forms of DDL, perioperative radiotherapy following National Comprehensive Cancer Network guidelines may also be considered.[8]
Retroperitoneal DDL is the most common, surgically unaccessible and serious form of DDL: it has a recurrence rate of 66% and a five-year overall survival rate of 54%.[31] The primary treatment option for retroperitoneal DDL is surgical resection. A phase III clinical trial found little difference in the results of radiation therapy followed by surgical resection compared to surgical resection alone in the treatment of retroperitoneal DDL.[6] In other phase III clinical trials, DDL patients with inaccessible retroperitoneal and/or metastatic tumors were treated with front-line chemotherapy comparing doxorubicin to doxorubicin plus ifosfamide or doxorubicin to gemcitabine plus docetaxel. Other studies have likewise examined the value of various chemotherapy regimens. These studies often found little difference in the overall survival times in their comparisons but did show some improvements in progression-free survival and other clinical parameters. Based on these studies, a recommended first-line therapy for retroperitoneal and other surgically inassessible or metastatic DDL tumors is treatment with an anthracycline-based chemotherapy regimen or, in tumor-resistant or relapsed cases, eribulin chemotherapy. A review conducted in 2020 reported median survival times for low histopathological grade and high histopathological grade DDL to be 113 months and 48 months, respectively.[32] Further studies are needed to provide evidence on the efficacies of radiotherapy, chemotherapy, and novel therapies in all the varieties of DDL.[33]
Novel therapies
Several novel therapy regimens for DDL and the more aggressive or otherwise problematic cases of ALT/WDL are currently undergoing clinical trials. A phase II clinical study investigating abemaciclib is underway in patients with pretreated or untreated DDL. Preliminary analysis showed that this inhibitor of the CDK4 and CDK6 genes' product Serine/threonine-specific protein kinase enzymes produced a prolonged median progression-free survival time of 30.4 weeks.[6] A phase III multicenter, randomized, double-blind, placebo-controlled clinical study of abemaciclib is in its active phase and will soon (as stated in July, 2021) begin recruiting 108 individuals with advanced, recurrent, and/or metastatic DDL. The study is sponsored by the Sarcoma Alliance for Research through Collaboration[34] in collaboration with Eli Lilly and Company.[35] Ribociclib, also a CDK4 and CDK6 gene inhibitor, in combination with a mTOR inhibitor, everolimus is in a phase II clinical trial in individuals with advanced DDL or leiomyosarcoma.[35] A phase III registration study (i.e. a large confirmatory study meant to establish an acceptable benefit/safety profile in order to gain regulatory approval for a precisely defined indication) is evaluating the safety and efficacy of milademetan compared to trabectedin in patients with unresectable (i.e., resection is deemed to cause unacceptable morbidity or mortality) or metastatic DDL that has progressed on 1 or more prior systemic therapies, including at least 1 anthracycline-based therapy. The sponsor, Rain Therapeutics Inc, is currently recruiting 160 individuals for the trial.[36] Another phase III clinical trial is investigating the MDM2 inhibitor milademetan[37] versus trabectedin, a blocker of the oncogenic transcription factor FUS-CHOP, in MDM2-overexpressing ALT/WDL and DDL.[38] Milademetan has shown manageable toxicity and some activity resulting in stable disease and/or a few partial responses in DDL.[6]
Myxoid liposarcoma
Presentation
Myxoid liposarcoma (MLS), which includes a type of liposarcoma termed round cell liposarcoma,[39] represents ~30% of all liposarcomas. It has a peak incidence in individuals' fourth and fifth decades with a male predominance in most studies. While uncommon in children and adolescents, MLS is the most common liposarcoma form diagnosed in these age groups. MLS typically presents as a large (1 to 39 cm; average 12 cm), mobile, well-circumscribed, painless mass that developed from 1 week to 15 years prior to diagnosis. MLS tumors are located in deep-seated soft tissues of the thighs (65–80% of cases), lower legs (10–15% of cases), retroperitoneum (8% of cases), and arms (5% of cases). In about one-third of cases, these tumors metastasize to other soft tissue sites (e.g. retroperitoneum, thorax, or other extremity), skeletal bone, and/or lung. Individuals may present with these metastasis, particularly those in bone; it has been recommended that patients should be tested at presentation for bone metastasis by medical imaging, including X-rays, CT scans, and/or magnetic resonance imaging.[40]
Pathology
Histopathologic analyses of MLS (see Figs. 3 and 4 in the below Histopathology of liposarcomas section) reveals cells scattered throughout a myxoid matrix (i.e. a connective tissue background that appears more blue or purple than the red color of normal connective tissue when these tissues are properly prepared, H&E stained, and viewed microscopically). These cells are lipoblasts, some of which are signet ring-shaped (a shape suggesting that the cell may be neoplastic), oval-shaped, or round-shaped.[40] MLS tumors may be hypercellular and contain solid sheets of round cells that comprise at least 5% of all cells or low cellularity populated with cells that have bland nuclei and <5% round cells in a background of curving capillaries resembling a chicken-wire pattern. Tumors that contain at least 5% round cells are classified as high-grade while those with <5% round cells are classified as low-grade.[39] High-grade MLS tumors typically take a more aggressive clinical course than low-grade MLS tumors.[40]
Genetics
MLS tumor cells are virtually defined by their expression of a FUS-DDIT3 fusion gene (also termed a chimeric gene) which occurs in >95% of cases or a EWSR1-DDIT3 fusion gene which occurs in the remaining <5% of cases. The FUS-DDIT3 fusion gene forms as a result of a translocation (termed t(12:16)(q13:p11)) between the site of the DDIT3 gene [41] at band 12 of chromosome 12's q arm and the site of the FUS gene[42] at band 11 on chromosome 16's short arm (also termed the p arm). The fusion protein (also termed chimeric protein) product of this chimeric oncogene gene, FUS-DDIT3, is known to arrest fat cell maturation and promote neoplasia. The EWSR1-DDIT3 fusion gene (termed t(12;22)(q13;q12)) results from a translocation of the EWSR1 gene[43] located at band 12.2 on chromosome 22's q arm with the DDIT2 gene. The fusion protein product of the EWSR1-DDIT3 gene, like the FUS-DDIT3 fusion protein, promotes neoplasia.[44] In spite of these fusion gene relations, further studies are required to define their contribution to the development and/or maintenance of MLS tumors.[6]
Diagnosis
Low-grade and intermediate-grade MLS tumors can be identified histologically by their classic morphology of distinctive chicken-wire vasculature scattered throughout a myxoid stroma. However, high-grade MLS tumors can be difficult to distinguish from other round cell neoplasms, particularly high grade MLS tumors that consist of diffuse cell and/or pure round cell morphology to such an extent as to obscure this classic vascular-myxoid pattern. Detection of a DDIT3 gene rearrangements with the FUS or EWSR1 gene by in situ hybridization or immunohistochemistry or the RNA fusion transcripts of these genes by real-time polymerase chain reactions confirms the diagnosis of high-grade as well as ambiguous cases of low-grade or intermediate-grade MLS tumors.[44]
Treatment and prognosis
MLS has typically been treated by surgical resection but may require more radical interventions, e.g. limb amputation may be needed when a limb's neurovascular bundle is compromised. The post-surgical risk of recurrence within 3 years after surgery has been reported to be ~15% when not all tumor is removed and ~10% when tumor removal is complete.[40] The addition of radiotherapy to surgical resection has improved the local control of MLS tumors and has been recommended to treat unresectable and recurrent MLS.[45] However, further studies are needed to determine the value of radiotherapy in treating the various varieties of MLS.[40] Chemotherapy regimens using ifosfamide, an anthracycline such as daunorubicin, dacarbazine, and/or trabectedin have been found useful: a phase III clinical trial showed progression-free survival times in MLS patients treated with trabectedin or dacarbazine to be 5.6 and 1.5 months, respectively. In 2015 the Food and Drug Administration approved trabectedin for use in unresectable and metastatic liposarcomas.[citation needed]
Overall, the 10-year survival rate of MLS individuals has been 77%, a survival rate appreciably longer than other liposarcoma forms. Compared to low-risk MLS, high-risk MLS (risk defined by tumor round cell content and/or other unfavorable prognostic indicators) is associated with increased rates of metastasis and therefore a shorter survival time. Increased tumor size (≥ 10 cm) is strongly associated with a higher grade MLS and therefore a shorter survival time. Other factors that have been associated with unfavorable outcomes in MLS include presence of tumor necrosis, age >45 years, P53 gene overexpression,[40] and male gender.[46] The round cell form of myxoid liposarcomas also appears to have a relatively poor prognosis: in various retrospective reviews, myxoid liposarcoma was usually found to be low-grade and therefore relatively responsive to chemotherapy whereas high grade (i.e. round cell) myxoid lipsarcoma had higher rates of metastasis, behaved more aggressively, and did not respond well to chemotherapy.[40] It is important to note, however, that almost all cases of myxoid liposarcomas in pediatric patients have had excellent prognoses.[45]
Novel therapies
A PPAR-γ agonist (i.e. activator), efatutazone,[47] was studied in a small phase I trial on individuals with various advanced-stage malignancies. The drug produced a markedly durable response in a person with MLS suggesting that PPAR-γ agonists would be useful for treating this disease.[48] A stage II clinical trial conducted in Italy is examining the effects of a trabectedin plus pioglitazone (another PPAR-γ agonist) in individuals with stable MLS tumors. The study involves two sequential steps. The first step examines the response of patients treated for a minimum of 4 cycles with trabectedin alone. If stable disease is attained, the second step will examine the effects of further treating initially responding patients with a combination of trabectedin and pioglitazone.[49] A stage II clinical trial is nearing completion to evaluate the efficacy of sirolimus (an inhibitor of MTOR; sirolimus is also known as rapamycin) plus cyclophosphamide (a chemotherapy drug) in metastatic or unresectable MLS.[50] A phase II clinical trial is recruiting patients to evaluate sintilimab (a human IgG4 monoclonal antibody directed against the programmed cell death protein 1 located on the surface of cells) in combination with two chemotherapy drugs, doxorubicin and ifosfamide, as first-line treatment of soft tissue sarcomas including MLS.[51]
T cells have been genetically engineered to target the MAGE-A4 antigen expressed on a HLA-A*02 MAGE-A4-containing peptide located on the surface of the neoplastic cells in certain types of tumors. These engineered cells (termed ADP-A2M4-T cells) attacked and killed various cultured human cancer cells bearing this antigen[52] and, in a clinical stage 1 study, shrank various solid tumor types in patients whose tumors' contained neoplastic cells expressing this antigen.[53] A phase II clinical study is now recruiting individuals to investigate the efficacy and safety of ADP-A2M4 T cells (engineered from the recipient's own T cells) in HLA-A*02-positive patients with metastatic or inoperable, advanced-stage MSGE-4-positive MLS tumors.[54]
Pleomorphic liposarcoma
Presentation
Pleomorphic liposarcomas (PLS), which account for 5% to 10% of all liposarcoma cases,[55] are fast-growing, usually large (>5 cm), and painless but highly malignant adipocyte tumors.[56] They occur primarily in individuals >50 years old[56] with a predominance in females.[45] PLS tumors are rarely found in children.[56] PLS tumors present in a leg or arm (65% of cases), retroperitoneum or abdomen (15% of cases),[6] or in rare cases the trunk wall, spermatic cord,[56] head and neck areas,[57] chest wall, pelvic cavity, pulmonary pleurae, pericardium, and spine.[55] These tumors are usually localized in deep soft tissues with only 25% of cases presenting in subcutaneous tissues.[56] Rare cases of PLS have presented in individuals with the Li-Fraumeni or Muir–Torre syndromes, two hereditary genetic disorders that predispose affected persons to develop various types of cancer.[45]
Pathology
The histopathology of PLS tumors often consists of areas resembling myxoid liposarcoma[58] mixed with areas containing undifferentiated cells.[56] These tumors are marked hypercellular and contain at least some variably shaped lipoblasts that have pleomorphic nuclei.[58] Areas of necrosis are common, giant cells, some of which are multinucleated and/or contain engulfed neutrophils, are occasionally present, and hyaline droplets may be seen in some cells as well as scattered extracellularly throughout the tumor.[59] The undifferentiated component of these tumors most often consists of spindle-shaped cells, with 25% of cases showing cells with an epithelioid cell morphology. These tumors have at least some foci with a histopathology similar to high-grade myxofibrosarcoma type histiocytomas,[56][58][60] a tumor formerly termed malignant myxoid fibrous histiocytoma.[61]
Genetics
PLS neoplastic cells contain various gene and chromosome abnormalities: the TP53 gene is deleted or mutated in 17–60% of cases; the RB1 gene is deleted in 60% of cases; and the Neurofibromin 1 gene is lost by inactivating mutations in 8% of cases or in rarer cases by a deletion around its location in band 11.2 on the long arm of chromosome 12. These cells can also show gains in the genetic material around: bands 12–15 on the short arm of chromosome 5; band 21 on the short arm of chromosome 1; and band 22 on the long arm of chromosome 7. The alterations in gene copy numbers induced by these abnormalities are similar to those seen in the myxofibrosarcoma type of the histiocytomas. The role(s) of these changes in gene copy numbers in promoting PLS has not been defined. Thus, PLS is unlike other liposarcomas in that its neoplastic cells have a complex genome without characteristic genomic alterations or identifiable genes that drive the disease. Detection of alterations in the expression of the TP53, RB1, and neurofibromin 1 genes, as well as other, less commonly altered genes in PLS (e.g. PIK3CA, tyrosine-protein kinase SYK, PTK2B, EPHA5, and ERBB4), may help support but do not clearly define a tumor as being PLS.[6][56] Extension of the chromosome telomere ends by pathological mechanisms termed alternative lengthening of telomeres occurs in the neoplastic cells of ~80% of PLS cases but is far less common or not seen in the other four forms of liposarcoma.[58]
Diagnosis
The diagnosis of PLS depends on its presentation, histopathology, and genetics. The histopathology of PLS often closely resembles that of myxofibrosarcoma but is distinguished from that tumor by its content of pleomorphic lipoblasts.[58]
Treatment and prognosis
Radical surgical resection is the main treatment for PLS; it is also an important palliative intervention to relieve symptoms due to the compression of organs and tissues. Surgery may require removal of an entire compressed organ such as the kidney or colon. Regardless of this surgery, however, local recurrence rates are very high. The uses of chemotherapy and/or radiotherapy in conjunction with radical surgery have not been shown to prolong survival and are regarded as controversial interventions.[55] The National Comprehensive Cancer Network recommends treatment for individuals with high-risk localized PLS by complete surgical resection, when feasible, combined with radiation therapy. Individuals with metastatic disease have been treated with chemotherapy (e.g. doxorubicin plus ifosfamide or eribulin) similar to the regimens used for dedifferentiated liposarcoma (see above section on the treatment of this liposarcoma type)[6] About 20% of PLS tumors metastasize to distant sites, the most common of which are lung (82% of metastases), liver (18% of metastases), and bone or pancreas (18% of metastases). PLS survival rates at 1, 3, and 5 years are reported to be 93%, 75%, and 29%, respectively. Tumors located in the center position of the trunk, larger than 10 cm in size, deeply seated, or containing areas of necrosis have worse prognoses.[55]
Myxoid pleomorphic liposarcoma
Myxoid pleomorphic liposarcoma (originally termed pleomorphic myxoid liposarcoma[62]) was first described in a large 2009 study of the liposarcomas.[63] While initially regarded as a possible variant of the myxoid liposarcomas with pleomorphic features, the World Health Organization (2020) classified it as a new and distinct form of the liposarcomas. This classification was based on findings that the myxoid pleomorphic liposarcomas, while having histopathological features that were similar to myxoid liposarcomas, had clinical and, most importantly, critical genetic and molecular features that differed from the myxoid as well as the other three liposarcoma forms.[5]
Presentation
Myxoid pleomorphic liposarcoma (MPL) is an exceptionally rare and highly aggressive form of the liposarcomas that develops in children, adolescents,[5] young adults,[6] and, in a more recent study, individuals >50 years old.[62] MPL tumors present as deep soft-tissue masses that are often located in the mediastinum[44] and, less often, the extremities, head and neck, abdominal cavity, or trunk.[6] At least two case of MPL have presented in individuals with the Li–Fraumeni syndrome, an inherited genetic disorder that predisposes individuals to develop various cancers.[58][64][65]
Pathology
On histopathologic analyses, MPL tumors consist of areas resembling conventional myxoid liposarcoma; these areas, which represent 30–50% of the total tumor areas, have an abundant myxoid matrix, a well-developed capillary vasculature, bland cells that are round and/or slightly spindle-shaped, vacuolated lipoblasts, and multinucleated cells shaped like small flowers. However, these areas also contain a scattering of highly pleomorphic cells that show greater degrees of nuclear enlargement and irregularity than the cells seen myxoid liposarcoma tumors. Other areas of MPL tumors are more cellular and consist of rapidly growing and highly pleomorphic lipoblasts.[62]
Genetics
The neoplastic cells in MPL do not express the FUS-DDIT3 or EWSR1-DDIT3 fusion genes that are expressed by the neoplastic cells in >95% or <5%, respectively, of myxoid fibrosarcoma cases.[62][6] Inactivation of the RB1 tumor suppressor gene due to its deletion or pathological suppression is found in all cases MPL. MPL neoplastic cells also commonly have other alterations in their chromosomes. They may show abnormal gains in some of the genetic material normally found on chromosomes 1, 6, 7, 8, 19, 21, and/or X and losses in the genetical material normally found on chromosomes 2, 3, 4, 5, 10, 11, 12, 13, 14, 15, 16, 17 and/or 22. The genetic material lost in band 14 on the long arm of chromosome 13 includes not only the RP1 gene but also the RCBTB2, DLEU1, and ITM2B genes. Due to its rarity and more recent definition, the molecular characteristics and importance of these genetic abnormalities have yet to be fully defined.[56] Nonetheless, studies have suggested that Losses in any one or more of the RB1, RCBTB2, DLEU1, and ITM2B genes, but particularly the RP1 gene, may be involved in contributing to the development and/or progression of MPL.[62]
Diagnosis
The diagnosis of MPL depends on its tumors clinical presentation, histopathological resemblance to myxoid liposarcoma, and, most critically, absence of the FUS-DDIT3 sn EWSR1-DDIT3 fusion genes in its neoplastic cells.[62][6]
Treatment and prognosis
While individuals with MPL have been treated with surgical resection to remove their tumors,[64][65][6][66] a 2021 review found that there were no consensus recommendations for the standard of care for MPL with respect to radiation and chemotherapy regimens (when used either alone or combined with surgery) for treating these tumors.[6]
Histopathology of liposarcomas

Fig. 1 Micrograph of bone formation in a liposarcoma tumor
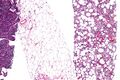
Fig. 2 Micrograph of a dedifferentiated liposarcoma tumor
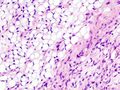
Fig. 3 Lower-power micrograph of myxoid liposarcoma tumor
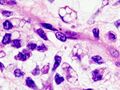
Fig. 4 Higher-power micrograph of myxoid liposarcoma tumor


.jpg)
.JPG)
Medical imaging
Medical ultrasonography and magnetic resonance imaging (MRI) of liposarcomas are helpful and often essential in determining their extent, surgical accessibility, and relationship to any observed organ dysfunctions. Since ultrasonography is usually unable to distinguish a liposarcoma from a benign lipoma, MRI is the initial imaging of choice to provide evidence relative to making this distinction.[67]
In myxoid liposarcoma, it shows low signal intensity mass with high signal intensity foci on T1-weighted MRI images. The mass shows high signal intensity on T2-weighted images. This is because it contains predominantly mucoid substance (accounts for low signal intensity on T1) and small amount of mature fat (accounts for high signal intensity on T1).[68] The mass is well-defined, lobulated, multiloculated, or oval in shape without any infiltration into surrounding structures.[68]

Fig. 5 Ultrasonography of a liposarcoma with high-echo areas reflected from its lipomatous matrix and low-echo areas reflected from its non-lipomatous areas.[69]
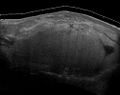
Fig. 6 Ultrasonography of a liposarcoma mimicking a lipoma. This homogeneous high-echoic mass has the same appearance as a lipoma.[69]

Fig. 7 MRI of myxoid liposarcoma of high grade, in the left axillary region of 40-year-old man, highlighted by its white color, in this horizontal section of the tumor.



Society and culture
Notable cases
- Chad Brown (1961–2014), a poker player, died from liposarcoma
- Richard Feynman (1918–1988), a theoretical physicist, died following surgery to address the disease.
- Rob Ford (1969–2016), former Toronto mayor and Toronto city councillor, died of pleomorphic liposarcoma.
- Hokie Gajan (1959–2016), former running back for the New Orleans Saints and radio color commentator for the team, died from liposarcoma.
- Charlie Davies (born 1986), former soccer player for the Philadelphia Union of Major League Soccer, diagnosed with liposarcoma in 2016.
- Mark Strand (1934–2014), former US Poet Laureate and Pulitzer Prize-winner, died from liposarcoma.
See also
- Lipoma
- The Wendy Walk, not-for-profit organization whose mission is to raise funds and awareness for sarcomas, including liposarcoma
References
- ↑ "Liposarcoma". https://www.pathologyoutlines.com/topic/softtissuewdliposarcoma.html. Topic Completed: November 2017. Minor changes: May 2023
- ↑ "Synchronous Renal Dedifferentiated Liposarcoma and Retroperitoneal Well-Differentiated Liposarcoma: A Case Report With Literature Review". International Journal of Surgical Pathology 29 (6): 667–671. December 2020. doi:10.1177/1066896920981682. PMID 33355009.
- ↑ Dei Tos AP (August 2000). "Liposarcoma: new entities and evolving concepts". Ann Diagn Pathol 4 (4): 252–66. doi:10.1053/adpa.2000.8133. PMID 10982304.
- ↑ Bell, Teresa (October 2012). "What is Liposarcoma?". http://sarcomahelp.org/liposarcoma.html.
- ↑ 5.0 5.1 5.2 5.3 5.4 5.5 5.6 5.7 "Update of pediatric soft tissue tumors with review of conventional MRI appearance-part 1: tumor-like lesions, adipocytic tumors, fibroblastic and myofibroblastic tumors, and perivascular tumors". Skeletal Radiology 51 (3): 477–504. June 2021. doi:10.1007/s00256-021-03836-2. PMID 34191084.
- ↑ 6.00 6.01 6.02 6.03 6.04 6.05 6.06 6.07 6.08 6.09 6.10 6.11 6.12 6.13 6.14 6.15 6.16 6.17 6.18 6.19 6.20 "Recent advances in the understanding and management of liposarcoma". Faculty Reviews 10: 1. 2021. doi:10.12703/r/10-1. PMID 33659920.
- ↑ "Contributions of cytogenetics and molecular cytogenetics to the diagnosis of adipocytic tumors". Journal of Biomedicine & Biotechnology 2011: 524067. 2011. doi:10.1155/2011/524067. PMID 21274402.
- ↑ 8.00 8.01 8.02 8.03 8.04 8.05 8.06 8.07 8.08 8.09 8.10 8.11 8.12 8.13 8.14 8.15 8.16 8.17 8.18 8.19 "Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review". Seminars in Diagnostic Pathology 36 (2): 112–121. March 2019. doi:10.1053/j.semdp.2019.02.006. PMID 30852045.
- ↑ "Dysplastic Lipoma: A Distinctive Atypical Lipomatous Neoplasm With Anisocytosis, Focal Nuclear Atypia, p53 Overexpression, and a Lack of MDM2 Gene Amplification by FISH; A Report of 66 Cases Demonstrating Occasional Multifocality and a Rare Association With Retinoblastoma". The American Journal of Surgical Pathology 42 (11): 1530–1540. November 2018. doi:10.1097/PAS.0000000000001129. PMID 30001242.
- ↑ "A Comprehensive Review on Solitary Fibrous Tumor: New Insights for New Horizons". Cancers 13 (12): 2913. June 2021. doi:10.3390/cancers13122913. PMID 34200924.
- ↑ 11.0 11.1 "Pathology and genetics of adipocytic tumors". Cytogenetic and Genome Research 118 (2–4): 138–47. 2007. doi:10.1159/000108294. PMID 18000364.
- ↑ 12.0 12.1 12.2 12.3 "Clinical Application of Chromosome Microarray Analysis in the Diagnosis of Lipomatous Tumors". Applied Immunohistochemistry & Molecular Morphology 29 (8): 592–598. March 2021. doi:10.1097/PAI.0000000000000923. PMID 33734108.
- ↑ "Atypical lipomatous tumor in the ligamentum teres of liver: A case report and review of the literature". World Journal of Clinical Cases 6 (12): 548–553. October 2018. doi:10.12998/wjcc.v6.i12.548. PMID 30397612.
- ↑ 14.0 14.1 14.2 14.3 "Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma of the Orbit: Three Cases and Review of the Literature". Ophthalmic Plastic and Reconstructive Surgery 37 (3S): S134–S140. 2021. doi:10.1097/IOP.0000000000001804. PMID 32991496.
- ↑ "MDM2 MDM2 proto-oncogene [Homo sapiens (Human)] - Gene - NCBI". https://www.ncbi.nlm.nih.gov/gene/4193.
- ↑ "CDK4 cyclin dependent kinase 4 [Homo sapiens (Human)] - Gene - NCBI". https://www.ncbi.nlm.nih.gov/gene/1019.
- ↑ 17.0 17.1 17.2 17.3 "Duplication of chromosome segment 12q13-15 in a lipomatous tumor with minimal nuclear atypia: A case report". Oncology Letters 11 (4): 2875–2878. April 2016. doi:10.3892/ol.2016.4305. PMID 27073568.
- ↑ 18.0 18.1 "Small supernumerary marker chromosomes (sSMC) and male infertility: characterization of five new cases, review of the literature, and perspectives". Journal of Assisted Reproduction and Genetics 37 (7): 1729–1736. July 2020. doi:10.1007/s10815-020-01811-9. PMID 32399795.
- ↑ 19.0 19.1 "Rare partial trisomy and tetrasomy of 15q11-q13 associated with developmental delay and autism spectrum disorder". Molecular Cytogenetics 13: 21. 2020. doi:10.1186/s13039-020-00489-z. PMID 32536972.
- ↑ "Liposarcoma". ESUN. 2004. http://sarcomahelp.org/liposarcoma.html.
- ↑ 21.0 21.1 "Primary Orbital Dedifferentiated Liposarcoma". World Neurosurgery 139: 604–607. July 2020. doi:10.1016/j.wneu.2020.04.069. PMID 32339743.
- ↑ "Osteogenic differentiation in dedifferentiated liposarcoma: a study of 36 cases in comparison to the cases without ossification". Histopathology 72 (5): 729–738. April 2018. doi:10.1111/his.13421. PMID 29076540.
- ↑ "Combining surgery with 125I brachytherapy for recurrent mediastinal dedifferentiated liposarcoma: A case report and review of literature". World Journal of Clinical Cases 8 (5): 939–945. March 2020. doi:10.12998/wjcc.v8.i5.939. PMID 32190631.
- ↑ "Meningothelial cells as part of the central nervous system host defence". Biology of the Cell 105 (7): 304–15. July 2013. doi:10.1111/boc.201300013. PMID 23634770.
- ↑ "Dedifferentiated Liposarcoma With Meningothelial-Like Whorls: Five Additional Cases and Review of the Literature". International Journal of Surgical Pathology 28 (7): 749–758. October 2020. doi:10.1177/1066896920921950. PMID 32419561.
- ↑ "MDM2 MDM2 proto-oncogene [Homo sapiens (Human)] – Gene – NCBI". https://www.ncbi.nlm.nih.gov/gene/4193.
- ↑ "CDK4 cyclin dependent kinase 4 [Homo sapiens (Human)] – Gene – NCBI". https://www.ncbi.nlm.nih.gov/gene/1019.
- ↑ "The Hidden Genomic and Transcriptomic Plasticity of Giant Marker Chromosomes in Cancer". Genetics 208 (3): 951–961. March 2018. doi:10.1534/genetics.117.300552. PMID 29279323.
- ↑ "YEATS4 YEATS domain containing 4 [Homo sapiens (Human)] – Gene – NCBI". https://www.ncbi.nlm.nih.gov/gene/8089.
- ↑ "The impact of chemotherapy on survival of patients with extremity and trunk wall soft tissue sarcoma: revisiting the results of the EORTC-STBSG 62931 randomised trial". European Journal of Cancer 109: 51–60. March 2019. doi:10.1016/j.ejca.2018.12.009. PMID 30690293.
- ↑ "Surgical Diagnosis and Treatment of Primary Retroperitoneal Liposarcoma". Frontiers in Surgery 8: 672669. 2021. doi:10.3389/fsurg.2021.672669. PMID 34150840.
- ↑ "Epidemiology and survival of liposarcoma and its subtypes: A dual database analysis". Journal of Clinical Orthopaedics and Trauma 11 (Suppl 4): S479–S484. July 2020. doi:10.1016/j.jcot.2020.04.013. PMID 32774015.
- ↑ "Role of Radiation Therapy for Newly Diagnosed Retroperitoneal Sarcoma". Current Treatment Options in Oncology 22 (9): 75. July 2021. doi:10.1007/s11864-021-00877-6. PMID 34213610.
- ↑ "Sarcoma Alliance for Research through Collaboration". https://sarcomaalliance.org/event/sarcoma-alliance-for-research-through-collaboration-2/.
- ↑ 35.0 35.1 "SARC041: Phase 3 Randomized Double-Blind Study of Abemaciclib Versus Placebo in Patients with Advanced Dedifferentiated Liposarcoma". 23 July 2021. https://clinicaltrials.gov/ct2/show/NCT04967521?term=abemaciclib&cond=Dedifferentiated+Liposarcoma&draw=2&rank=1.
- ↑ "A Randomized Multicenter Phase 3 Study of Milademetan Versus Trabectedin in Patients with Dedifferentiated Liposarcoma". 16 July 2021. https://clinicaltrials.gov/ct2/show/NCT04979442?cond=Dedifferentiated+Liposarcoma&draw=2&rank=1.
- ↑ "Milademetan". https://pubchem.ncbi.nlm.nih.gov/compound/Milademetan?form=MY01SV&OCID=MY01SV.
- ↑ "A Randomized Multicenter Phase 3 Study of Milademetan Versus Trabectedin in Patients with Dedifferentiated Liposarcoma". 16 July 2021. https://clinicaltrials.gov/ct2/show/NCT04979442?term=mdm2&cond=Dedifferentiated+Liposarcoma&draw=2&rank=1.
- ↑ 39.0 39.1 Zafar, R.; Wheeler, Y. (2021). "Liposarcoma". StatPearls. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK538265/.
- ↑ 40.0 40.1 40.2 40.3 40.4 40.5 40.6 "Myxoid Liposarcoma With Skeletal Metastases: Pathophysiology and Imaging Characteristics". Current Problems in Diagnostic Radiology 50 (1): 66–73. 2021. doi:10.1067/j.cpradiol.2019.10.008. PMID 31813645.
- ↑ "DDIT3 DNA damage inducible transcript 3 [Homo sapiens (Human)] – Gene – NCBI". https://www.ncbi.nlm.nih.gov/gene/1649.
- ↑ "FUS FUS RNA binding protein [Homo sapiens (Human)] – Gene – NCBI". https://www.ncbi.nlm.nih.gov/gene/2521.
- ↑ "EWSR1 EWS RNA binding protein 1 [Homo sapiens (Human)] – Gene – NCBI". https://www.ncbi.nlm.nih.gov/gene/2130.
- ↑ 44.0 44.1 44.2 "Nuclear expression of DDIT3 distinguishes high-grade myxoid liposarcoma from other round cell sarcomas". Modern Pathology 34 (7): 1367–1372. July 2021. doi:10.1038/s41379-021-00782-1. PMID 33731886.
- ↑ 45.0 45.1 45.2 45.3 "Adipocytic tumors in Children: A contemporary review". Seminars in Diagnostic Pathology 36 (2): 95–104. March 2019. doi:10.1053/j.semdp.2019.02.004. PMID 30850231.
- ↑ "Molecular signatures of tumor progression in myxoid liposarcoma identified by N-glycan mass spectrometry imaging". Laboratory Investigation 100 (9): 1252–1261. September 2020. doi:10.1038/s41374-020-0435-2. PMID 32341520.
- ↑ "Efatutazone dihydrochloride". https://www.wikidata.org/wiki/Q27266481.
- ↑ "A Phase II Study of the Peroxisome Proliferator-Activated Receptor Gamma Agonist, Efatutazone in Patients with Previously Treated, Unresectable Myxoid Liposarcoma". 16 August 2021. https://clinicaltrials.gov/ct2/show/study/NCT02249949?term=myxoid+liposarcoma&draw=2&rank=7.
- ↑ "A Phase II Study on Trabectedin in Combination with PPARg Agonist Pioglitazone in Patients with Round Cell Myxoid Liposarcomas or Dedifferentiated G1 and G2 Liposarcomas with Stable Disease After a Monotherapy with Trabectedin. (TRABEPIO)". 10 March 2021. https://clinicaltrials.gov/ct2/show/NCT04794127?term=myxoid+liposarcoma&draw=2&rank=4.
- ↑ "A Phase 2, Single Arm, Multi Center Trial Evaluating the Efficacy of the COmbination of Sirolimus and cYclophosphamide in Metastatic or Unresectable Myxoid Liposarcoma and chOndrosarcoma". 4 June 2021. https://clinicaltrials.gov/ct2/show/NCT02821507?cond=Myxoid+Liposarcoma&draw=2&rank=6.
- ↑ Luo, Zhiguo (13 July 2020). "A Single Arm, Multi Centers, Phase II Study of Sintilimab, Doxorubicin and Ifosfamide at First-line Treatment of Soft Tissue Sarcoma Including Undifferentiated Pleomorphic Sarcoma, Synovial Sarcoma, Myxoid Liposarcoma and De-differentiated Liposarcoma". https://clinicaltrials.gov/ct2/show/NCT04356872?recrs=ab&cond=myxoid+liposarcoma&draw=2&rank=4.
- ↑ "Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy". Oncoimmunology 9 (1): 1682381. 2020. doi:10.1080/2162402X.2019.1682381. PMID 32002290.
- ↑ "T Cells Targeting MAGE-A4 Shrink Tumors". Cancer Discovery 10 (8): OF2. August 2020. doi:10.1158/2159-8290.CD-NB2020-059. PMID 32540953.
- ↑ "A Phase 2 Single Arm Open-Label Clinical Trial of ADP-A2M4 SPEAR™ T Cells in Subjects with Advanced Synovial Sarcoma or Myxoid/Round Cell Liposarcoma". 18 June 2021. https://clinicaltrials.gov/ct2/show/NCT04044768?cond=Myxoid+Liposarcoma&draw=2&rank=5.
- ↑ 55.0 55.1 55.2 55.3 "Pleomorphic liposarcoma: An analysis of 6 case reports and literature review". Medicine 97 (8): e9986. February 2018. doi:10.1097/MD.0000000000009986. PMID 29465602.
- ↑ 56.0 56.1 56.2 56.3 56.4 56.5 56.6 56.7 56.8 "The Rapidly Expanding Group of RB1-Deleted Soft Tissue Tumors: An Updated Review". Diagnostics 11 (3): 430. March 2021. doi:10.3390/diagnostics11030430. PMID 33802620.
- ↑ "Pleomorphic liposarcoma of the head and neck: Presentation of two cases and literature review". American Journal of Otolaryngology 38 (4): 505–507. 2017. doi:10.1016/j.amjoto.2017.04.012. PMID 28528729.
- ↑ 58.0 58.1 58.2 58.3 58.4 58.5 "Molecular updates in adipocytic neoplasms✰". Seminars in Diagnostic Pathology 36 (2): 85–94. March 2019. doi:10.1053/j.semdp.2019.02.003. PMID 30857767.
- ↑ "Pleomorphic liposarcoma". https://www.pathologyoutlines.com/topic/softtissueadiposepleomorphiclipo.html.
- ↑ "Integrated genetic and epigenetic analysis of myxofibrosarcoma". Nature Communications 9 (1): 2765. July 2018. doi:10.1038/s41467-018-03891-9. PMID 30018380. Bibcode: 2018NatCo...9.2765O.
- ↑ "Myxofibrosarcoma of the leg: A diagnostic challenge". Clinical Case Reports 8 (12): 3333–3336. December 2020. doi:10.1002/ccr3.3414. PMID 33363928.
- ↑ 62.0 62.1 62.2 62.3 62.4 62.5 "Myxoid pleomorphic liposarcoma-a clinicopathologic, immunohistochemical, molecular genetic and epigenetic study of 12 cases, suggesting a possible relationship with conventional pleomorphic liposarcoma". Modern Pathology 34 (11): 2043–2049. June 2021. doi:10.1038/s41379-021-00862-2. PMID 34168281.
- ↑ Alaggio, Rita; Coffin, Cheryl M.; Weiss, Sharon W.; Bridge, Julia A.; Issakov, Josephine; Oliveira, Andre M.; Folpe, Andrew L. (May 2009). "Liposarcomas in Young Patients: A Study of 82 Cases Occurring in Patients Younger Than 22 Years of Age". American Journal of Surgical Pathology 33 (5): 645–658. doi:10.1097/PAS.0b013e3181963c9c. PMID 19194281.
- ↑ 64.0 64.1 "Recurrent Pleomorphic Myxoid Liposarcoma in a Patient With Li-Fraumeni Syndrome". International Journal of Surgical Pathology 28 (2): 225–228. April 2020. doi:10.1177/1066896919878804. PMID 31559875.
- ↑ 65.0 65.1 "Pleomorphic myxoid liposarcoma in an adolescent with Li-Fraumeni syndrome". Pediatric Surgery International 33 (5): 631–635. May 2017. doi:10.1007/s00383-017-4063-x. PMID 28160093.
- ↑ "Liposarcoma in children and young adults: a clinicopathologic and molecular study of 23 cases in one of the largest institutions of China". Virchows Archiv 479 (3): 537–549. March 2021. doi:10.1007/s00428-021-03076-8. PMID 33738541.
- ↑ Rohit Sharma; Frank Gaillard. "Lipoma". https://radiopaedia.org/articles/lipoma.
- ↑ 68.0 68.1 Sung, Mi-Sook; Kang, Heong S.; Suh, Jin S.; Lee, Jung H.; Park, Jeong M.; Kim, Jee Y.; Lee, Hae G. (July 2000). "Myxoid Liposarcoma: Appearance at MR Imaging with Histologic Correlation" (in en). RadioGraphics 20 (4): 1007–1019. doi:10.1148/radiographics.20.4.g00jl021007. ISSN 0271-5333. PMID 10903690. http://pubs.rsna.org/doi/10.1148/radiographics.20.4.g00jl021007.
- ↑ 69.0 69.1 Content originally copied from: Mak, Chee-Wai; Tzeng, Wen-Sheng (2012). "Sonography of the Scrotum". Sonography. doi:10.5772/27586. ISBN 978-953-307-947-9. https://www.intechopen.com/books/sonography/sonography-of-the-scrotum. under the CC-BY-3.0 license.
| Classification | |
|---|---|
| External resources |
 |  |

