Chemistry:Nitrile
In organic chemistry, a nitrile is any organic compound that has a –C≡N functional group. The name of the compound is composed of a base, which includes the carbon of the –C≡N, suffixed with "nitrile", so for example CH
3CH
2C≡N is called "propionitrile" (or propanenitrile).[1] The prefix cyano- is used interchangeably with the term nitrile in industrial literature. Nitriles are found in many useful compounds, including methyl cyanoacrylate, used in super glue, and nitrile rubber, a nitrile-containing polymer used in latex-free laboratory and medical gloves. Nitrile rubber is also widely used as automotive and other seals since it is resistant to fuels and oils. Organic compounds containing multiple nitrile groups are known as cyanocarbons.
Inorganic compounds containing the –C≡N group are not called nitriles, but cyanides instead.[2] Though both nitriles and cyanides can be derived from cyanide salts, most nitriles are not nearly as toxic.
Structure and basic properties
The N−C−C geometry is linear in nitriles, reflecting the sp hybridization of the triply bonded carbon. The C−N distance is short at 1.16 Å, consistent with a triple bond.[3] Nitriles are polar, as indicated by high dipole moments. As liquids, they have high relative permittivities, often in the 30s.
History

The first compound of the homolog row of nitriles, the nitrile of formic acid, hydrogen cyanide was first synthesized by C. W. Scheele in 1782.[4][5] In 1811 J. L. Gay-Lussac was able to prepare the very toxic and volatile pure acid.[6]
Around 1832 benzonitrile, the nitrile of benzoic acid, was prepared by Friedrich Wöhler and Justus von Liebig, but due to minimal yield of the synthesis neither physical nor chemical properties were determined nor a structure suggested. In 1834 Théophile-Jules Pelouze synthesized propionitrile, suggesting it to be an ether of propionic alcohol and hydrocyanic acid.[7] The synthesis of benzonitrile by Hermann Fehling in 1844 by heating ammonium benzoate was the first method yielding enough of the substance for chemical research. Fehling determined the structure by comparing his results to the already known synthesis of hydrogen cyanide by heating ammonium formate. He coined the name "nitrile" for the newfound substance, which became the name for this group of compounds.[8][9]
In 1903, Arthur Lapworth investigated the formation of cyanohydrins by addition of hydrocyanic acid to aldehydes and ketones and discovered that the actual nucleophile is the cyanide ion, such that the addition of a base increases the reaction rate. This work represented one of the earliest investigations of an organic reaction mechanism.[10][11]
For a long time, nitriles were primarily of academic interest. Between the First and Second World War, however, research activity increased significantly.[8] By the second half of the 20th century, several large-scale industrial processes had been developed in which nitriles were either produced or utilized. An important example is the development of polyamides (polyamide 6.6) in the 1930s, as adiponitrile is a key intermediate in its manufacture and is produced by hydrocyanation of butadiene with hydrogen cyanide.[12][13] Acrylonitrile polymers have been known since the 1920s but gained greater importance as synthetic fibers toward the late 1940s.[14] Superglues based on cyanoacrylates have also been available since the late 1940s.[15]
Nomenclature
according to IUPAC: Butanenitrile (blue marked C atom belongs to the main chain),
formally also propanecarbonitrile (blue marked C atom belongs to the substituent)
The functional group of nitriles containing the C≡N triple bond is referred to as the nitrile or cyano group.[16] If the nitrile is the highest-ranking functional group, the suffix -nitrile is added to the name of the parent compound. The triply bonded carbon atom is, as always, included in the parent chain.[17] Alternatively, the ending -carbonitrile may be used (analogous to -carboxylic acid), in which case the carbon atom is not counted as part of the parent chain.[18] This ending must be used if the nitrile group is attached to a ring (as in cyclopentanecarbonitrile ({{{2}}})) or if not all carbon atoms are part of the parent chain, which is necessarily the case when more than two nitrile groups are present, as these can only be located at the termini of the chain.[19] Due to their relationship to carboxylic acids (the nitrile carbon has the same oxidation state as the carboxyl carbon), trivial names are often derived from the corresponding carboxylic acids using the ending -onitrile (for example, benzoic acid to benzonitrile).[20] If the nitrile function is not the principal functional group in the molecule, the prefix cyano- is used together with the appropriate locant. In this case as well, the triple-bonded carbon atom is not counted as part of the parent chain.[19]
Synthesis
Numerous methods are available for the preparation of nitriles. These include Kolbe nitrile synthesis, dehydration of carboxylic acid amides and oximes, and oxidation of primary amines.
Industrially, the main methods for producing nitriles are ammoxidation and hydrocyanation. Both routes are green in the sense that they do not generate stoichiometric amounts of salts.
From organic halides and cyanide salts
Two salt metathesis reactions are popular for laboratory scale reactions. In the Kolbe nitrile synthesis, alkyl halides undergo nucleophilic aliphatic substitution with alkali metal cyanides. Aryl nitriles are prepared in the Rosenmund-von Braun synthesis.
In general, metal cyanides combine with alkyl halides to give a mixture of the nitrile and the isonitrile, although appropriate choice of counterion and temperature can minimize the latter. An alkyl sulfate obviates the problem entirely, particularly in nonaqueous conditions (the Pelouze synthesis).[5]
In the Kolbe nitrile synthesis (a nucleophilic substitution reaction), an alkanonitrile and an alkali halide are formed from a reactive halocarbons and an alkali cyanide (sodium cyanide or potassium cyanide). The reaction is particularly suitable for primary, allylic, and benzylic halides. Secondary alkyl halides provide lower yields, whereas tertiary halides undergo exclusively elimination reaction instead of substitution. In addition to halides, substrates bearing other good leaving groups may also be employed. In contrast to alkali cyanides, silver cyanide is unsuitable for nitrile synthesis, as it preferentially forms isonitriles.[21] An example of the Kolbe nitrile synthesis is the reaction of methyl iodide with sodium cyanide to yield acetonitrile and sodium iodide:[22]
Similarly, 1,3-dibromopropane reacts with sodium cyanide to form glutaronitrile[23], and 1-iodooctane reacts with potassium cyanide to give nonannitrile.[24] Cyanations can also be carried out using hydrogen cyanide in combination with triethylaluminum or with diethylaluminum cyanide; for example, in the ring opening of an epoxide to a β-cyanohydrin or in the 1,4-addition of cyanide to an enone.[25][26] Trimethylsilylcyanide is another cyanating reagent capable of opening epoxides to β-cyanohydrins, with concomitant silylation of the oxygen atom.[27] Trimethylsilyl cyanide also enables substitution of tertiary alkyl halides, which is not feasible under Kolbe nitrile synthesis conditions.[21]
In the presence of suitable transition metal catalysts, hydrocyanation allows addition of hydrogen cyanide to the multiple bonds of alkenes and alkynes to afford nitriles. Nickel catalysts are typically employed. Direct handling of hydrogen cyanide is often unnecessary, as synthetic equivalents such as acetone cyanohydrin or isovaleronitrile may be used.[28] An important industrial process is the hydrocyanation of butadiene to adiponitrile.[13]
Hydrocyanation
Hydrocyanation is an industrial method for producing nitriles from hydrogen cyanide and alkenes. The process requires homogeneous catalysts. An example of hydrocyanation is the production of adiponitrile, a precursor to nylon-6,6 from 1,3-butadiene:
- CH
2=CH–CH=CH
2 + 2 HC≡N → NC(CH
2)
4C≡N
Dehydration of amides and others
Nitriles can be prepared by the dehydration of primary amides. Common reagents for this include phosphorus pentoxide (P
2O
5)[29] and thionyl chloride (SOCl
2).[30] In a related dehydration, secondary amides give nitriles by the von Braun amide degradation. In this case, one C-N bond is cleaved.

Specifically, carboxamides and oximes can be converted to nitriles by dehydration (elimination of water). Numerous reagents and methodologies are available for this transformation.[31][32][33] Methods for nitrile synthesis via dehydration of nitroalkanes have also been described.[34]
Phosphorus pentoxide, known since the mid-19th century, is a classical reagent for amide dehydration.[31] Amides can also be dehydrated using trivalent phosphorus reagents such as phosphorus trichloride or triphenyl phosphite;[33] as well as diethyl chlorophosphate,[35] thionyl chloride[36], or phosgene.[37] In the presence of specific palladium complexes or other suitable catalysts, acetonitrile can function as a dehydrating agent, converting an amide into a nitrile while being transformed into acetamide. Similarly, dichloroacetonitrile may be employed.[38][39] Related systems utilize iron(II) chloride tetrahydrate, zinc trifluoromethanesulfonate, or uranyl nitrate as catalysts in combination with N-methyl-N-trimethylsilyltrifluoroacetamide as the dehydrating reagent.[40][41][42] Carboxylic acid amides can also be dehydrated using a system comprising triphenylphosphane, iodine, and 4-methylmorpholine.[43] Another approach involves high-temperature dehydration (220–240 °C) in hexamethylphosphoramide (HMPA).[44] Dehydration of primary amides with zinc chloride under microwaves is reversible. In aqueous acetonitrile, an amide can be converted to a nitrile; however, in a water–tetrahydrofuran system with added acetamide, the reverse conversion of nitrile to amide occurs.[45]
Both carboxamides and aldoximes can be dehydrated using aluminum chloride and sodium iodide in acetonitrile.[32] Likewise, both classes can be dehydrated with oxalyl chloride and catalytic dimethyl sulfoxide in a reaction analogous to the Swern oxidation.[46] Conversion to nitriles under catalysis by heptavalent rhenium species (perrhenic acid or trimethylsilyl perrhenate) is effective for both amides and aldoximes; the water formed can be removed by azeotropic distillation.[47]
The conversion of aldehydes to nitriles via aldoximes is a popular laboratory route. Aldehydes react readily with hydroxylamine salts, sometimes at temperatures as low as ambient, to give aldoximes. These can be dehydrated to nitriles by simple heating,[48] although a wide range of reagents may assist with this, including triethylamine/sulfur dioxide, zeolites, or sulfuryl chloride. The related hydroxylamine-O-sulfonic acid reacts similarly.[49]
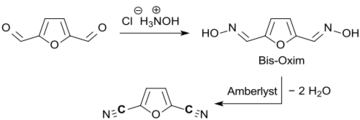
One-pot synthesis from aldehyde (Amberlyst is an acidic ion-exchange resin.)

In specialised cases the Van Leusen reaction can be used. Biocatalysts such as aliphatic aldoxime dehydratase are also effective.
Aldoximes may also be dehydrated with cyanuric chloride,[50] the Burgess reagent,[51] or a combination of trifluoromethanesulfonic acid anhydride and triphenylphosphine, the latter being oxidized to triphenylphosphine oxide.[52] Catalytic dehydrogenation is likewise possible, for example with iron(III) triflate ({{{2}}}),[53] copper(II) acetate,[54] mixed hydroxides of tin and tungsten,[55] or a bimetallic palladium–manganese catalyst.[56] Enzymatic dehydration of aldoximes using aldoxime dehydratases has also been achieved. These bacterial enzymes, including those from Pseudomonas chlororaphis, have been applied repeatedly in nitrile synthesis.[57]
Preparation from aldehydes and ketones
Aldehydes can be converted into oximes using hydroxylamine hydrochloride and subsequently dehydrated to nitriles (e.g., with oxalyl chloride).[58] Direct transformation of aldehydes to nitriles is also possible using hydroxylamine-O-sulfonic acid[59] or O-(4-trifluoromethylbenzoyl)hydroxylamine ({{{2}}}).[60] Such conversions can also be accomplished with hydroxylamine in the presence of titanium(IV) chloride or mixed tin–tungsten hydroxides as catalysts[55][61], or by addition of sulfuryl fluoride or selenium dioxide.[62][63] Tosylmethylisocyanide (Van Leusen reagent) enables direct conversion of ketones into nitriles via the Van Leusen reaction, introducing the entire nitrile group and thus an additional carbon atom.[64][65][66]
Oxidation of primary amines

Numerous traditional methods exist for nitrile preparation by amine oxidation.[67] Common methods include the use of potassium persulfate,[68] Trichloroisocyanuric acid,[69] or anodic electrosynthesis.[70] In addition, several selective methods have been developed in the last decades for electrochemical processes.[71]
Several procedures employ nitroxyl radicals such as TEMPO or 4-acetamido-TEMPO as catalytic oxidants. These catalysts can be regenerated either by potassium peroxymonosulfate as the stoichiometric oxidant or electrochemically under applied potential.[72][73] Another approach utilizes copper(I) chloride or copper(II) chloride as catalyst, molecular oxygen as the stoichiometric oxidant, and a molecular sieve to remove the water formed.[74]
Ammoxidation
In ammoxidation, a hydrocarbon is partially oxidized in the presence of ammonia. This conversion is practiced on a large scale for acrylonitrile:[75]
- 2 CH
3CH=CH
2 + 3 O
2 + 2 NH
3 → 2 N≡CCH=CH
2 + 6 H
2O
In the production of acrylonitrile, a side product is acetonitrile. On an industrial scale, several derivatives of benzonitrile, phthalonitrile, as well as Isobutyronitrile are prepared by ammoxidation. The process is catalysed by metal oxides and is assumed to proceed via the imine.
Ammoxidation is a heterogeneously catalyzed gas-phase reaction in which aliphatic or methyl-substituted aromatic compounds react with oxygen (air) and ammonia to form nitriles, with water as a by-product. Reaction temperatures exceed 300 °C, and oxides of vanadium, chromium, or molybdenum serve as catalysts.[76] Acrylonitrile, an important precursor for polymer production (see Use section), is primarily manufactured by ammoxidation of propene.[14] The principal industrial route to hydrogen cyanide is the Andrussov process, i.e., ammoxidation of methane over a platinum catalyst. However, a significant proportion of global hydrogen cyanide production arises as a by-product of acrylonitrile manufacture.[77]
Preparation of aromatic nitriles

Aryl nitriles can be synthesized via the Sandmeyer reaction of diazonium salts with copper(I) cyanide[78] or by the Rosenmund-von Braun reaction (direct reaction of an aryl bromide with copper(I) cyanide).[79] Conversion of thiocyanate with aromatic carboxylic acids, known as Letts nitrile synthesis, can be carried out using potassium thiocyanate; lead thiocyanate generally provides higher yields.[8]
Aryl iodides can be converted into aromatic nitriles under palladium catalysis with trimethylsilyl cyanide. For example, iodobenzene reacts with trimethylsilyl cyanide in the presence of tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) to form benzonitrile.[80] Another palladium-catalyzed route (also employing Pd(PPh3)4) is the decarbonylation of aromatic acyl cyanides.[81] Palladium-catalyzed cyanation of aryl chlorides with potassium cyanide[82] or potassium hexacyanidoferrate(II)[83] has likewise been reported. Quinones can react with trimethylsilyl cyanide to give silylated cyanohydrins, which are subsequently aromatized using phosphorus tribromide.[84] A further approach involves reaction of aryl Grignard or aryllithium reagents with dimethylmalonitrile.[85]
Aromatic nitriles are often prepared in the laboratory from the aniline via diazonium compounds. This is the Sandmeyer reaction. It requires transition metal cyanides.[86]
- ArN+
2 + CuC≡N → ArC≡N + N
2 + Cu+
Preparation of cyanohydrins
The cyanohydrins are a special class of nitriles. Classically they result from the addition of alkali metal cyanides to aldehydes in the cyanohydrin reaction. Because of the polarity of the organic carbonyl, this reaction requires no catalyst, unlike the hydrocyanation of alkenes. O-Silyl cyanohydrins are generated by the addition trimethylsilyl cyanide in the presence of a catalyst (silylcyanation). Cyanohydrins are also prepared by transcyanohydrin reactions starting, for example, with acetone cyanohydrin as a source of HCN.[87]
Cyanohydrins can also be prepared by addition of an alkali cyanide to an aldehyde or ketone in the presence of acetic acid. For less reactive substrates, diethylaluminum cyanide provides a suitable alternative. Another approach is transhydrocyanation, in which hydrogen cyanide is transferred from acetone cyanohydrin to an aldehyde or ketone.[11] Suitable catalysts for this transformation include lanthanide alkoxides such as lanthanum(III) isopropoxide ({{{2}}}), cerium(III) isopropoxide ({{{2}}}), samarium(III) isopropoxide ({{{2}}}), and ytterbium(III) isopropoxide ({{{2}}}).[88]
Addition of trimethylsilyl cyanide to aldehydes or ketones affords cyanohydrins as their trimethylsilyl ethers.[89][90] Suitable catalysts include zinc iodide, potassium cyanide in combination with 18-crown-6, or ytterbium(III) cyanide.[11] Under appropriate conditions, such reactions can be rendered enantioselective. Vanadium- or titanium-based catalysts bearing chiral salen-type ligands are suitable, as is the combination of tetraisopropyl orthotitanate with a chiral imine.[91][92]
Preparation of acyl cyanides
Acyl cyanides (α-oxonitriles) can in certain cases be prepared by reacting carboxylic acid halides with transition metal cyanides (e.g., copper cyanide or silver cyanide). This approach is particularly effective for aromatic carboxylic acid halides and aliphatic acyl bromides, whereas aliphatic acyl chlorides are unreactive. Aliphatic acyl cyanides can instead be synthesized by reacting carboxylic acid chlorides with trimethylsilyl cyanide.[93]
Enantioselective synthesis of chiral nitriles
Using chiral pool starting materials, enantioselective synthesis enables access to α-chiral nitrile-containing compounds in eutomeric form, such as vildagliptin and saxagliptin. Conventional transformations can introduce the nitrile functionality; for example, an enantiomerically pure amide or oxime derived from naturally enantiopure proline may be dehydrated. The applicability of such strategies depends on the specific target molecule. Asymmetric cyanation reactions are also established.[94] Of particular importance is the asymmetric hydrocyanation of carbonyl compounds (see section on cyanohydrin preparation). In addition, numerous asymmetric hydrocyanations of imines have been developed, affording enantiomerically pure α-aminonitriles.[92]
Other methods
- A commercial source for the cyanide group is diethylaluminum cyanide Et
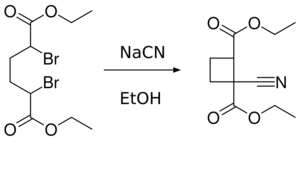
2AlCN which can be prepared from triethylaluminium and HCN.[95] It has been used in nucleophilic addition to ketones.[96] For an example of its use see: Kuwajima Taxol total synthesis - Cyanide ions facilitate the coupling of dibromides. Reaction of α,α′-dibromoadipic acid with sodium cyanide in ethanol yields the cyano cyclobutane:[97]
- Aromatic nitriles can be prepared from base hydrolysis of trichloromethyl aryl ketimines (RC(CCl
3)=NH) in the Houben-Fischer synthesis[98] - α-Amino acids form nitriles and carbon dioxide via various means of oxidative decarboxylation.[99][100] Henry Drysdale Dakin discovered this oxidation in 1916.[101]
- From aryl carboxylic acids (Letts nitrile synthesis)

- Carbocyanation enables addition of a nitrile group across a multiple bond to yield a further nitrile. Aryl nitriles can be added to alkynes under catalysis by bis(cyclooctadiene)nickel(0) and trimethylphosphine, affording α,β-unsaturated nitriles. Modification of the reaction conditions, for example by employing a different phosphane or adding a frustrated Lewis pair such as trimethylaluminum or triphenylborane, allows addition of non-aromatic nitriles, both saturated and α,β-unsaturated.[102] Carbocyanation reactions that couple two molecules while introducing a nitrile group are also known, using hexabutyldistannane and tosyl cyanide as the cyanide source.[103]
- Carboxylic acids can be converted to the corresponding nitriles by reaction with indium(III) chloride in acetonitrile at 200 °C. In this process, acetonitrile functions both as solvent and nitrogen source and is converted into acetic acid. The reaction proceeds via multiple Mumm rearrangements.[104] Alcohols can be transformed into nitriles by a Mitsunobu reaction, employing cyanomethylidene trimethyl phosphorane in the presence of acetone cyanohydrin.[105] N-Alkylamides can be converted to nitriles via the von Braun degradation using phosphorus pentachloride.[106] Alternative reagents include phosphorus pentabromide and carbonyl bromide.[107]
Reactions
Nitrile groups in organic compounds can undergo a variety of reactions depending on the reactants or conditions. A nitrile group can be hydrolyzed, reduced, or ejected from a molecule as a cyanide ion.
Hydrolysis
The hydrolysis of nitriles RCN proceeds in the distinct steps under acid or base treatment to first give carboxamides RC(O)NH
2 and then carboxylic acids RC(O)OH. The hydrolysis of nitriles to carboxylic acids is efficient. In acid or base, the balanced equations are as follows:
- RC≡N + 2 H
2O + HCl → RC(O)OH + NH
4Cl - RC≡N + H
2O + NaOH → RC(O)ONa + NH
3
Strictly speaking, these reactions are mediated (as opposed to catalyzed) by acid or base, since one equivalent of the acid or base is consumed to form the ammonium or carboxylate salt, respectively.
Kinetic studies show that the second-order rate constant for hydroxide-ion catalyzed hydrolysis of acetonitrile to acetamide is 1.6×10−6 M−1 s−1, which is slower than the hydrolysis of the amide to the carboxylate (7.4×10−5 M−1 s−1). Thus, the base hydrolysis route will afford the carboxylate (or the amide contaminated with the carboxylate). On the other hand, the acid catalyzed reactions requires a careful control of the temperature and of the ratio of reagents in order to avoid the formation of polymers, which is promoted by the exothermic character of the hydrolysis.[108] The classical procedure to convert a nitrile to the corresponding primary amide calls for adding the nitrile to cold concentrated sulfuric acid.[109] The further conversion to the carboxylic acid is disfavored by the low temperature and low concentration of water.
- RC≡N + H
2O → RC(O)NH
2
Two families of enzymes catalyze the hydrolysis of nitriles. Nitrilases hydrolyze nitriles to carboxylic acids:
- RC≡N + 2 H
2O → RC(O)OH + NH
3
Nitrile hydratases are metalloenzymes that hydrolyze nitriles to amides.
- RC≡N + H
2O → RC(O)NH
2
These enzymes are used commercially to produce acrylamide.
The "anhydrous hydration" of nitriles to amides has been demonstrated using an oxime as water source:[110]
- RC≡N + R'C(H)=NOH → RC(O)NH
2 + R'C≡N
Reduction
Nitriles are susceptible to hydrogenation over diverse metal catalysts. The reaction can afford either the primary amine (RCH
2NH
2) or the tertiary amine ((RCH
2)
3N), depending on conditions.[111] In conventional organic reductions, nitrile is reduced by treatment with lithium aluminium hydride to the amine. Reduction to the imine followed by hydrolysis to the aldehyde takes place in the Stephen aldehyde synthesis, which uses stannous chloride in acid.
Deprotonation
Alkyl nitriles are sufficiently acidic to undergo deprotonation of the C-H bond adjacent to the C≡N group.[112][113] Strong bases are required, such as lithium diisopropylamide and butyl lithium. The product is referred to as a nitrile anion. These carbanions alkylate a wide variety of electrophiles. Key to the exceptional nucleophilicity is the small steric demand of the C≡N unit combined with its inductive stabilization. These features make nitriles ideal for creating new carbon-carbon bonds in sterically demanding environments.
Nucleophiles
The carbon center of a nitrile is electrophilic, hence it is susceptible to nucleophilic addition reactions:
- with an organozinc compound in the Blaise reaction
- with alcohols in the Pinner reaction.
- with amines, e.g. the reaction of the amine sarcosine with cyanamide yields creatine[114]
- with arenes to form ketones in the Houben–Hoesch reaction via an imine intermediate.
- with Grignard reagents to form primary ketimines in the Moureau-Mignonac ketimine synthesis.[115] While not a classical Grignard reaction, it may be considered one under broader modern definitions.
Miscellaneous methods and compounds
- In reductive decyanation the nitrile group is replaced by a proton.[116] Decyanations can be accomplished by dissolving metal reduction (e.g. HMPA and potassium metal in tert-butanol) or by fusion of a nitrile in KOH.[117] Similarly, α-aminonitriles can be decyanated with other reducing agents such as lithium aluminium hydride.[116]
- In the so-called Franchimont Reaction (developed by the Belgian doctoral student Antoine Paul Nicolas Franchimont (1844-1919) in 1872), an α-cyanocarboxylic acid heated in acid hydrolyzes and decarboxylates to a dimer.[118]
- Nitriles self-react in presence of base in the Thorpe reaction in a nucleophilic addition
- In organometallic chemistry nitriles are known to add to alkynes in carbocyanation:[119]

Complexation
Nitriles are precursors to transition metal nitrile complexes, which are reagents and catalysts. Examples include tetrakis(acetonitrile)copper(I) hexafluorophosphate ([Cu(MeCN)
4]+
) and bis(benzonitrile)palladium dichloride (PdCl
2(PhCN)
2).[120]
Nitrile derivatives
Nitriles are isomeric with isonitriles (isocyanides). These also contain a C≡N triple bond; however, the substituent is bonded via the nitrogen atom, which results in a zwitterionic structure.[121]
Compounds in which an oxygen atom is bonded to the carbon atom of a C≡N group are referred to as cyanates.[122] If the oxygen atom is replaced by a sulfur or selenium atom, the compounds are termed thiocyanates or selenocyanates ({{{2}}}).[123][124] If the cyano group is bonded to a nitrogen atom, the compound is referred to as a cyanamide.[125]
In addition to nitriles, other classes of compounds are known that contain a C≡N triple bond in which the nitrogen atom forms a fourth bond and is therefore positively charged. In nitrile oxides, an oxygen atom is additionally bonded to the nitrogen atom.[126] If this atom is sulfur or another nitrogen atom instead, the compounds are referred to as nitrile sulfides ({{{2}}}) or nitrilimines, respectively.[127][128] If the nitrogen atom of the nitrile is protonated or carries an additional organic substituent, the compound is a nitrilium ion.[129] If the nitrogen atom carries an organic substituent bearing a negatively charged carbon atom, the species is a nitrile ylide, a subclass of ylides.[130]
Organic cyanamides
Cyanamides are N-cyano compounds with general structure R1
R2
N–C≡N and related to the parent cyanamide.[131]
Nitrile oxides
Nitrile oxides have the chemical formula RCNO. Their general structure is R–C≡N+
–O−
. The R stands for any group (typically organyl, e.g., acetonitrile oxide CH
3–C≡N+
–O−
, hydrogen in the case of fulminic acid H–C≡N+
–O−
, or halogen (e.g., chloroformonitrile oxide ({{{2}}}) Cl–C≡N+
–O−
).[132]: 1187–1192
Nitrile oxides are quite different from nitriles and do not arise from direct oxidation of the latter.[133] Instead, they can be synthesised by nitroalkane dehydration, oxime dehydrogenation,[134]: 934–936 or halooxime elimination in base.[135] They are highly reactive in 1,3-dipolar cycloadditions,[132]: 1187–1192 such as to isoxazoles,[134]: 1201–1202 and undergo type I dyotropic rearrangement to isocyanates.[132]: 1700
The heavier nitrile sulfides are extremely reactive and rare, but temporarily form during the thermolysis of oxathiazolones. They react similarly to nitrile oxides.[136]
Occurrence
More than 100 naturally occurring nitriles were known as early as the 1990s,[137] and several hundred have since been identified.[138] These compounds occur in bacteria, fungi, plants, and arthropods and sponges.[137][138] The biosynthesis of naturally occurring nitriles frequently begins with amino acids. Their N-hydroxylation followed by decarboxylation (cleavage of the carboxylic acid group as carbon dioxide) yields oximes, which serve as the direct precursors of nitriles.[137]
Occurrence in plants
Numerous nitriles occur as secondary metabolites in plants.
In Ricinus communis (Ricinus communis), in addition to the highly toxic protein ricin, the alkaloid ricinin is present, which contains a nitrile functional group.[139] The structurally closely related nudiflorin occurs in Trevia nudiflora (family spurge family).[140] In brown mustard, indoleacetonitrile is present; it is formed from indoleacetaldoxime and presumably functions in defense against pathogenic fungi.[141] In jojoba, various nitriles are found, including simmondsin, a glycoside containing an α,β-unsaturated nitrile moiety in the aglycone.[142] A similar compound, menis daurin, occurs in European holly (Ilex aquifolium).[143] α,β-Unsaturated nitriles are also present in several species of the genus Acacia, including Sutherlandin and Acacipetalin.[144][145] The horseradish tree (horseradish tree) contains niazirine, a glycoside of 4-hydroxyphenylacetonitrile.[146] The fragrant sweet pea (Lathyrus odoratus) causes the disease lathyrism, for which N-glutamyl-3-aminopropionitrile and its degradation product 3-aminopropionitrile are responsible.[147][148] The essential oil of Heracleum transcaucasicum (genus hogweed) contains geranylnitrile.[149] 3-cyanopyridine is found in annual bindweed.[150] Cyanolipids are a class of lipids that occur exclusively in soap tree plants (Sapindaceae). Their alcohol component is an unsaturated nitrile with five carbon atoms and one or two hydroxy groups, in contrast to glycerol in glycerides. Soap tree plants containing cyanolipids include soapnut tree and guarana.[151][152] Hydrogen cyanide is released by many plants containing corresponding cyanogenic compounds, particularly cyanogenic glycosides and cyanolipids.[153] In plants, hydrogen cyanide also functions as a signaling molecule.[154]
-
Ricinus plant
-
Ricinin
-
Guaraná, a soap tree plant, contains cyanolipids
-
An alcohol component of cyanolipids, found for example in guaraná
Nitriles from glucosinolates in cruciferous plants
An important group of natural products that serve as precursors of nitriles are the mustard oil glycosides (glucosinolates), which are biosynthesized analogously to direct nitrile formation via an aldoxime intermediate.[137] Glucosinolates constitute a major class of secondary metabolites produced by plants of the cruciferous family (Brassicaceae) for defense against herbivores and microorganisms. Normally, glucosinolates are hydrolyzed by myrosinase to isothiocyanates; however, in the presence of an additional protein (epithio specifier protein), nitriles are formed instead.[155][156] Sinigrin is found primarily in horseradish, wasabi, and brown mustard, but also in head cabbage, kale, cauliflower, and Brussels sprouts; in addition to allyl isothiocyanate, it can be degraded to allyl cyanide (3-butenenitrile).[157][158] Glucotropaeolin, present in garden cress, is degraded to phenylacetonitrile; gluconasturtiin, found in watercress, is degraded to 3-phenylpropionitrile.[159] Sinalbin, occurring in Lepidium draba, can analogously be degraded to 4-hydroxyphenylacetonitrile.[160]
-
Watercress
-
 Structure of gluconasturtiin
Structure of gluconasturtiin -
Structure of phenylpropionitrile

Cyanohydrins and cyanogenic glycosides
Cyanohydrins and their glycosides, referred to as cyanogenic glycosides, are widespread in nature and occur in several thousand plant species.[11][138] More than one hundred naturally occurring cyanogenic glycosides have been identified.[138] Plants utilize cyanogenic glycosides for defense and possibly also as a nitrogen storage buffer. They are biosynthesized from a limited number of amino acids and various carbohydrates.[11] Upon tissue damage, the glycosides come into contact with enzymes (Β-glucosidase and hydroxynitrillyase), which first release the aglycone (a cyanohydrin) and subsequently cleave it into a carbonyl compound and toxic hydrocyanic acid. Amygdalin is a glycoside of mandelonitrile and one of the most widespread cyanogenic glycosides; it occurs particularly in the seeds of the rose family (Rosaceae), including cultivated apple, apricot, peach, plum, cherry, and almond tree.[161] Whereas amygdalin is confined to the seeds of peaches, other parts of the plant predominantly contain prunasin.[162] Prunasin is likewise a glycoside of mandelonitrile; however, its sugar moiety is a monosaccharide (rather than a disaccharide as in amygdalin). In almonds and bitter almonds, prunasin serves as a biosynthetic precursor of amygdalin.[163] Prunasin is also present in laurel cherry.[164] Prunasin and sambunigrin, along with several other cyanogenic glycosides, occur in passion flower; in papaya, prunasin predominates.[165][166] Sambunigrin, also a glycoside of mandelonitrile, is found in several species of the genus elderberry (Sambucus), including black elderberry and Canadian elderberry,[167][168] as well as in Ximenia americana.[169] Vicianin, another mandelonitrile glycoside, occurs in ferns of the genus Davellia (family Davalliaceae).[170] Dhurrin is a cyanogenic glycoside of 4-hydroxymandelonitrile found in sorghum millet and other species of the genus sorghum millet, including Sorghum halepense.[171][172] Linamarin (with the aglycone acetone cyanohydrin) and lotaustralin (with the aglycone butanone cyanohydrin) occur in the genera Linum (for example in common flax) and lotus flowers, as well as in the common bean.[173] Both compounds are also present in cassava.[174] The mistletoe species Loranthus micranthus (genus Loranthus) contains linamarin gallate, a derivative in which linamarin is additionally esterified with gallic acid.[175] The rubber tree also contains linamarin; studies indicate that in this case the compound likely serves as an important storage substance in addition to its defensive function. The seeds contain particularly high concentrations, and during seedling development the compound is metabolized without releasing hydrocyanic acid, suggesting utilization in other biosynthetic pathways.[176]
-
 Peach tree
Peach tree -
 Structure of amygdalin
Structure of amygdalin -
 Struktur des Prunasins
Struktur des Prunasins -
Mandelonitrile, the aglycone of amygdalin and prunasin



Occurrence in animals
Numerous arthropods (Arthropoda) contain cyanogenic (hydrogen cyanide-releasing) nitrile compounds, including centipedes (Chilopoda), bipedes (Diplopoda), beetles (Hemiptera), beetles (Coleoptera), and butterflies (Lepidoptera).[177] The gooseberry moth (Abraxas grossulariata) contains the nitrile-bearing glycoside sarmentosin, which likely functions in defense.[178] Sarmentosin is also present in several species of the genus Parnassius.[179] Several species of glass-winged bug (including Jadera haematoloma) contain cyanolipids or cardiospermine, which they may acquire from their host plants through sequestration of toxins, i.e., uptake and storage.[177][180][181] Six-spotted damselflies are butterflies capable of both sequestering the cyanogenic glycosides linamarin and lotaustralin from their host plants and synthesizing them de novo.[182] Other species of the same genus (Zygaena), such as the Marsh Hornwort, also contain cyanogenic glycosides.[183] The defensive secretion of the centipede Himantarium gabrielis contains benzoyl cyanide, phenylacetonitrile, mandelonitrile (benzaldehyde cyanohydrin), and mandelonitrile benzoate.[184] Phenylacetonitrile also functions as a hormone in the desert locust (Schistocerca gregaria).[185] In various tapeworms (Polydesmida), the defensive secretion likewise contains benzoyl cyanide.[186] The mite species Oribatula tibialis (order horn mite, Oribatida) contains mandelonitrile hexanoate.[187] Hydrogen cyanide also occurs in arthropods as a degradation product of cyanogenic compounds.[154]
-
Gooseberry moth
-
 Desert locust
Desert locust -
 Himantarium gabrielis
Himantarium gabrielis -
Benzoyl cyanide is found in the defensive secretions of various arthropods


In addition to arthropods, marine animals also contain nitrile compounds. These include bursatellin from broad-footed snails of the genus Bursatella[188] and the calyculins isolated from sponges.[189] The albanitriles from sponges of the genus Mycale are linear compounds (chain length 16 to 18 carbon atoms) bearing a nitrile group at one or both termini and several additional C≡C triple bonds.[190]
Occurrence in fungi
Many fungi produce hydrogen cyanide from glycine. These include representatives of the genera funnel mushrooms (Clitocybe), dwindlers (Marasmius), stem porcini (Polyporus), and Ritterlinge (Tricholoma).[191] The epurpurins are a group of yellow phenolic pigments, each bearing two nitrile groups, occurring in Emericella purpurea.[192] Diatretin II occurs in Fleshy Fungus (Clitocybe diatreta)[193] and in the purple reddish bolete.[194] In the clove dwarf mushroom, the cyanohydrin of glyoxylic acid is present; it is formed from two glycine molecules and releases hydrocyanic acid upon tissue damage.[195]
Occurrence in bacteria
Hydrogen cyanide is produced by various soil bacteria, including cyanobacteria and representatives of the genera Aeromonas, Bacillus, and Pseudomonas. Biosynthesis proceeds from glycine.[196] A group of alkanenitriles was isolated from Pseudomonas veronii: dodecannitrile, tridecannitrile, tetradecanenitrile, pentadecannitrile, and hexadecannitrile, as well as compounds of similar chain length containing a double bond. From Micromonospora echinospora, structurally related compounds were also isolated, differing by a terminal methyl branch, a double bond, or both.[197] A cyanohydrin containing a phosphonic acid moiety is known from Streptomyces regensis.[198] The aetokthonotoxin from the cyanobacteria Aetokthonos hydrillicola is a brominated indole derivative bearing a nitrile group. It is a potent neurotoxin that frequently causes mortality in bald eagles that ingest it.[199]
Occurrence in space
Nitriles are among the most abundant organic molecules in space, and more than ten distinct compounds have been unequivocally detected.[200] Hydrogen cyanide was one of the first polyatomic species identified in space and occurs there relatively frequently and in substantial quantities.[201] Other nitriles detected in space include acetonitrile and aminoacetonitrile,[200] as well as butyronitrile,[202] cyanoacetylene, and cyanopolyins containing two to five conjugated triple bonds.[203] Hydrogen cyanide, cyanoacetylene, and cyanogen are present in the atmosphere of Saturn's moon Titan.[204]
Significance for the origin of life
Nitriles may have played a significant role in chemical evolution on Earth.[205][204] Experimental studies have demonstrated that hydrogen cyanide can form under a wide range of plausible prebiotic conditions. Possible starting materials include gas mixtures of methane, carbon dioxide, nitrogen, ammonia, and/or hydrogen. Various energy sources, such as electrical discharges or ultraviolet radiation, are likewise conceivable. Under simple conditions, hydrogen cyanide can give rise to numerous additional organic molecules.[206] Hydrogen cyanide and other nitriles, such as cyanoacetylene and dicyan, are considered potential precursors of nucleic bases.[204][206] Aminonitriles, in turn, are regarded as likely precursors of amino acids and peptides; for example, aminoacetonitrile is a precursor of glycine. An analogous process to the Strecker synthesis is proposed, in which α-aminopropionitrile initially forms from cyanide, acetaldehyde, and ammonia and is subsequently hydrolyzed to alanine.[205][207][208]
Use
Hydrogen cyanide is used on a large scale in the chemical industry as an intermediate for the production of other compounds. Acrylonitrile is an important feedstock for the manufacture of nitrile polymers. Acetonitrile is an important solvent. Other nitriles are employed as fragrances, pesticides, and chemical reagents. The nitrile group also plays a significant role in the development of active pharmaceutical ingredients.
Use of hydrogen cyanide
Hydrogen cyanide is a bulk chemical; global production in 2001 was approximately 2.6 million tons. Key derivatives produced from it include adiponitrile, acetone cyanohydrin, sodium cyanide, and cyanuric chloride.[77][209] Chelating agents are also synthesized from hydrogen cyanide,[209] for example ethylenediaminetetraacetic acid from formaldehyde, ethylenediamine, hydrogen cyanide, and sodium hydroxide.[210] An important industrial route to amino acids is the Strecker synthesis, in which hydrogen cyanide serves as a starting material.[211] A quantitatively important amino acid is methionine, which is produced from acrolein, hydrogen cyanide, and hydrogen sulphide and is used primarily in animal feed.[209][212]
Plastics production
Several widely used polymers contain acrylonitrile as a monomer and therefore incorporate nitrile groups. Pure polyacrylonitrile (PAN) is difficult to process; consequently, during its production, 85 to 99% acrylonitrile is almost always copolymerized with small amounts of other monomers.[14] Copolymers containing 35 to 85% acrylonitrile, together with other monomers such as vinyl acetate and methyl methacrylate, are also employed.[14][213] Nitrile polymers are among the most important fully synthetic materials for textile fibers, alongside polyesters and polyamidess.[14][214] These fibers, known as acrylic fibers, are produced on the scale of several million tons per year. In 2000, global production was approximately 2.7 million tons.[14][215] Acrylic fibers are used in garments (such as socks and sweaters), blankets, carpets, and knitting yarn, among other applications.[14][216] PAN is also the principal precursor for the production of carbon fiber, which is used as an exceptionally lightweight yet strong material in automotive and aircraft construction.[14][213][217][218] Global production of the monomer acrylonitrile was approximately 3.2 million tons in 1988.[77]
Acrylonitrile butadiene rubbers are known as nitrile rubbers and exhibit advantageous properties such as high tensile strength, high abrasion resistance, and resistance to hydrocarbons (oils and fuels). They are therefore used for sealing rings and for oil and fuel hoses.[14] Another important application of nitrile rubber is protective gloves, which are frequently used in healthcare instead of latex clothing gloves, as the latter often cause latex allergies.[219] Such gloves are also commonly used when handling hazardous chemicals, including organic solvents.[220]
Another important polymer is the terpolymer of acrylonitrile, butadiene, and styrene (acrylonitrile-butadiene-styrene copolymer). This material is widely used for the outer housings of electronic devices (computers, monitors, and keyboards).[221] Other applications include automotive plastic components (e.g., headlight and mirror housings), refrigerator liners, housings for kitchen appliances, vacuum cleaners, and power tools, as well as suitcases, snack containers,[222] and toys, including Lego.[223][224] ABS is also produced on the scale of several million tons annually; for example, about 2.7 million tons were manufactured in 1992.[222]
Polyamide (nylon) is not a nitrile polymer; however, a key intermediate in its production is adiponitrile. Adiponitrile is obtained by hydrocyanation of butadiene or by dimerization of acrylonitrile and is converted by catalytic hydrogenation into hexamethylenediamine, one of the monomers used to produce nylon. The second monomer, adipic acid, is produced by oxidation of cyclohexane.[225][226] Acetone cyanohydrin is an important intermediate in the production of methyl methacrylate, which in turn is used to manufacture polymethyl methacrylate.[227]
-
Polyacrylonitrile is used in knitting yarn
-
Carbon fibers are often made from polyacrylonitrile
-
Protective gloves made from nitrile rubber
-
 Lego bricks are made from acrylonitrile butadiene styrene copolymer (ABS)
Lego bricks are made from acrylonitrile butadiene styrene copolymer (ABS)

Chemical-pharmaceutical industry and laboratories
Acetonitrile is used as a solvent, particularly in the pharmaceutical industry.[228] According to a market analysis, approximately 180,000 tons of acetonitrile were produced worldwide in 2022, of which around 70% was consumed by the pharmaceutical sector.[229] It is also one of the most important solvents for analyses performed by high-performance liquid chromatography.[228][230] The thermal decomposition of azobisisobutyronitrile (AIBN) and related compounds (e.g., azobis(cyclohexanecarbonitrile)) generates relatively stable radicals; accordingly, these compounds are used as radical initiators in radical reactions, particularly polymerizations.[231] The quinone DDQ, which contains two nitrile groups, is a widely used oxidizing agent, including in pharmaceutical synthesis.[232] Nitrile groups can be incorporated into biomolecules as probes for infrared spectroscopic investigations.[233] Some nitriles serve as starting materials for the synthesis of pharmaceuticals.[234] Ketoprofen is an anti-inflammatory agent approved in some EU countries; propionitrile is used in its industrial synthesis.[235][236]
Nitriles in medicine
Nitriles occur in numerous classes of drugs. Between 2010 and 2020, at least one drug containing a nitrile function was approved annually by the US Food and Drug Administration. The nitrile group exhibits characteristic physicochemical properties that are important in drug design. Structurally, it has a linear geometry and occupies very little space—approximately one eighth of the volume of a methyl group. As a ligand substituent, it is therefore well suited to occupying narrow and deep cavities within the binding site of a target protein that are otherwise difficult to access. Incorporation of a nitrile group into a molecule generally reduces its octanol-water partition coefficient or increases its aqueous solubility. This often favorably influences bioavailability, plasma half-life, and thus the duration of action of lipophilic compounds. In medicinal products, the nitrile group is typically metabolically stable.[237] The nitrile group is isosteric with the carbonyl group, the hydroxy group, and the chlorine atom. It therefore exhibits similar electronic and steric properties and can be exchanged with these groups to fine-tune molecular characteristics.[238]
The hydrogen bond represents the principal pharmacodynamic interaction of the nitrile group, which acts as a proton acceptor due to the electronegativity of its nitrogen atom, in contrast to the ethynyl group.[239] For example, the nitrile group of the competitive PDE-3 inhibitor milrinone forms an affinity-relevant hydrogen bond via a histidine residue located at the binding site of these phosphodiesterases.[237] Nitriles can form a coordinative bond with calcium cations, which is essential for the activity of calcium antagonists of the verapamil type. These agents inhibit calcium influx by forming, through ligand–calcium complex chemistry, a salt bridge with one of the glutamic acid residues in the selectivity filter within the pore of the calcium channel.[240][241] Verapamil is used in cardiovascular diseases such as arterial hypertension and angina pectoris.[242]
Nitrile substituents decrease the electron density of aromatic compounds through a strong inductive effect. In this manner, π-π interactions between a drug molecule and suitable amino acid residues of a target protein, such as phenylalanine, tyrosine, tryptophan, and histidine, are modulated.[238] Such π-π interactions are observed with the aromatase inhibitors letrozole and anastrozole, which act as antiestrogens and are used in breast cancer.[243][244] Many androgen receptor antagonists contain a markedly electron-deficient aromatic ring, which is particularly important for supramolecular receptor binding.[245] In bicalutamide, enzalutamide, and other analogs used to treat prostate cancer, a nitrile group contributes to this electronic effect.[246]
In some cases, nitriles form a reversible yet pharmacologically relevant covalent bond with a target molecule.[238] Under appropriate conditions, an addition reaction can occur between serine or cysteine residues of the target protein and the nitrile group to form imidic acid esters or thioimidates. This mechanism applies to the dipeptidyl peptidase 4 inhibitor vildagliptin used in diabetes mellitus,[247] as well as to saxagliptin.[248] The antibacterial antibiotic cefmetazole also acts as a covalent inhibitor, in this case targeting a bacterial peptidase.[249] The calcium sensitizer levosimendan is presumed to react with the cardiac troponin protein complex.[250] Such a reactive functional group is also referred to as a warhead.
In certain cases, nitrile groups exert their effect primarily through steric interactions (i.e., spatial complementarity) by forming van der Waals forces with amino acid residues. This applies to the tyrosine kinase inhibitor bosutinib, which is used in chronic myeloid leukemia. Crystal structures have been reported in which bosutinib is complexed with various tyrosine kinases. Inhibitors of reverse transcriptase, such as Etravirin and Rilpivirin, are used in combination therapies against HIV. The acrylonitrile substructure of rilpivirine penetrates an aromatic cage composed of tyrosine, phenylalanine, and tryptophan, as demonstrated by the corresponding three-dimensional structure published in 2008.[251] The serotonin reuptake inhibitor citalopram, used in the treatment of depression, was the most frequently prescribed psychotropic drug in Germany in 2016, with 290 million defined daily doses. The nitrile group of escitalopram exhibits optimal complementarity to both the central and an additional allosteric binding site of the transporter protein, as evidenced by crystal structure analysis.[252]
-
Levosimendan
-
 Letrozole
Letrozole -
 Saxagliptin
Saxagliptin -
 Milrinone
Milrinone -
 Cefmetazole
Cefmetazole -
Citalopram
-
 Rilpivirine
Rilpivirine





Pharmaceuticals
Over 30 nitrile-containing pharmaceuticals are currently marketed for a diverse variety of medicinal indications with more than 20 additional nitrile-containing leads in clinical development. The types of pharmaceuticals containing nitriles are diverse, from vildagliptin, an antidiabetic drug, to anastrozole, which is the gold standard in treating breast cancer. In many instances the nitrile mimics functionality present in substrates for enzymes, whereas in other cases the nitrile increases water solubility or decreases susceptibility to oxidative metabolism in the liver.[253] The nitrile functional group is found in several drugs.
-
 Structure of periciazine, an antipsychotic studied in the treatment of opiate dependence
Structure of periciazine, an antipsychotic studied in the treatment of opiate dependence -
 Structure of citalopram, an antidepressant drug of the selective serotonin reuptake inhibitor (SSRI) class
Structure of citalopram, an antidepressant drug of the selective serotonin reuptake inhibitor (SSRI) class -
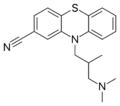 Structure of cyamemazine, an antipsychotic drug
Structure of cyamemazine, an antipsychotic drug -
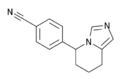 Structure of fadrozole, an aromatase inhibitor for the treatment of breast cancer
Structure of fadrozole, an aromatase inhibitor for the treatment of breast cancer -
 Structure of letrozole, an oral nonsteroidal aromatase inhibitor for the treatment of certain breast cancers
Structure of letrozole, an oral nonsteroidal aromatase inhibitor for the treatment of certain breast cancers




Other uses
A few dozen nitriles are used as fragrance ingredients in cosmetics. These include cinnamic acid nitrile, dodecanitrile, benzonitrile, and geranyl nitrile.[254][255][256] Some nitriles possess fragrances similar to those of the corresponding aldehydes but are considerably more stable, making them suitable substitutes. For example, geranyl nitrile provides a citrus note and, unlike the structurally analogous citral, is resistant to oxidation.[257]
Various nitriles are employed as pesticides. Cyano groups are present in certain pyrethroids. Pyrethroids are carboxylic acid esters; by using 3-phenoxymandelonitrile as the alcohol component, as in deltamethrin and cypermethrin, a class of particularly potent derivatives has been developed.[258] Azoxystrobin became the world's best-selling agricultural fungicide in 1999, only a few years after its introduction, with sales exceeding 400 million US dollars, and it has retained its market significance for more than 15 years, remaining the leading fungicide in 2016.[259][260] Azoxystrobin was developed on the basis of the naturally occurring strobilurin. Key structural modifications relative to the parent compound include replacement of double bonds with aromatic rings and introduction of a cyano group onto the pre-existing ring system.[260] A widely used nitrile-containing insecticide is fipronil.[261]
cyanoacrylates are used as adhesives because, as single-component formulations, they cure rapidly under ambient conditions and can bond a wide range of materials. By far the most widely used compound in this field is 2-cyanoacrylic acid ethyl ester, while 2-cyanoacrylic acid methyl ester and allyl cyanoacrylate are used to a lesser extent.[262] Cyanoacrylate adhesives are also applied in medicine for wound closure as an alternative to suturing. However, short-chain alkyl esters (e.g., methyl cyanoacrylate) frequently cause adverse effects, particularly inflammation; therefore, different compounds are employed in medical applications than in technical uses. In particular, butyl cyanoacrylate and 2-octyl cyanoacrylate are predominantly used.[15]
Nitriles are used as electrolyte additives in lithium batterys. For example, the addition of 1,3,6-hexanetricarbonitrile leads to a significant performance improvement compared with a corresponding battery without such an additive. The mechanism of action of nitrile additives has not yet been fully elucidated.[263][264]
See also
- Protonated nitriles: Nitrilium
- Deprotonated nitriles: Nitrile anion
- Cyanocarbon
- Nitrile ylide
References
- ↑ IUPAC Gold Book nitriles
- ↑ NCBI-MeSH Nitriles
- ↑ Karakida, Ken-ichi; Fukuyama, Tsutomu; Kuchitsu, Kozo (1974). "Molecular Structures of Hydrogen Cyanide and Acetonitrile as Studied by Gas Electron Diffraction". Bulletin of the Chemical Society of Japan 47 (2): 299–304. doi:10.1246/bcsj.47.299.
- ↑ See:
- Carl W. Scheele (1782) "Försök, beträffande det färgande ämnet uti Berlinerblå" (Experiment concerning the colored substance in Berlin blue), Kungliga Svenska Vetenskapsakademiens handlingar (Royal Swedish Academy of Science's Proceedings), 3: 264–275 (in Swedish).
- Reprinted in Latin as: "De materia tingente caerulei berolinensis" in: Carl Wilhelm Scheele with Ernst Benjamin Gottlieb Hebenstreit (ed.) and Gottfried Heinrich Schäfer (trans.), Opuscula Chemica et Physica (Leipzig ("Lipsiae"), (Germany): Johann Godfried Müller, 1789), vol. 2, pages 148–174.
- ↑ 5.0 5.1 David T. Mowry (1948). "The Preparation of Nitriles". Chemical Reviews 42 (2): 189–283. doi:10.1021/cr60132a001. PMID 18914000.
- ↑ Gay-Lussac produced pure, liquified hydrogen cyanide in: Gay-Lussac, J (1811). ""Note sur l'acide prussique" (Note on prussic acid)". Annales de chimie 44: 128–133. https://books.google.com/books?id=uJs5AAAAcAAJ&pg=PA128.
- ↑ J. Pelouze (1834). "Notiz über einen neuen Cyanäther". Annalen der Pharmacie 10 (3): 249. doi:10.1002/jlac.18340100302. https://books.google.com/books?id=P0s9AAAAcAAJ&pg=PA249.
- ↑ 8.0 8.1 8.2 David T. Mowry (1948-04-01), "The Preparation of Nitriles.", Chemical Reviews 42 (2): 189–283, doi:10.1021/cr60132a001, PMID 18914000
- ↑ Hermann Fehling (1844). "Ueber die Zersetzung des benzoësauren Ammoniaks durch die Wärme (On the decomposition of ammonium benzoate by heat)". Annalen der Chemie und Pharmacie 49 (1): 91–97. doi:10.1002/jlac.18440490106. https://books.google.com/books?id=2U09AAAAcAAJ&pg=PA91. On page 96, Fehling writes: "Da Laurent den von ihm entdeckten Körper schon Nitrobenzoyl genannt hat, auch schon ein Azobenzoyl existirt, so könnte man den aus benzoësaurem Ammoniak entstehenden Körper vielleicht Benzonitril nennen." (Since Laurent named the substance that was discovered by him "nitrobenzoyl" – also an "azobenzoyl" already exists – so one could name the substance that originates from ammonium benzoate perhaps "benzonitril".)
- ↑ Arthur Lapworth (1903), "XCVI.—Reactions involving the addition of hydrogen cyanide to carbon compounds", J. Chem. Soc., Trans. 83: 995–1005, doi:10.1039/CT9038300995
- ↑ 11.0 11.1 11.2 11.3 11.4 Robert J. H. Gregory (1999-12-08), "Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications", Chemical Reviews 99 (12): 3649–3682, doi:10.1021/cr9902906, PMID 11849033, Bibcode: 1999ChRv...99.3649G
- ↑ Encyclopedia of chemical processing and design. 2: Additives to alpha, New York: Dekker, 1977, ISBN 0-8247-2452-6
- ↑ 13.0 13.1 Ji Yang, Peng Wang, Helfried Neumann, Ralf Jackstell, Matthias Beller (2023), "Industrially applied and relevant transformations of 1,3-butadiene using homogeneous catalysts", Industrial Chemistry & Materials 1 (2): 155–174, doi:10.1039/D3IM00009E
- ↑ 14.0 14.1 14.2 14.3 14.4 14.5 14.6 14.7 14.8 "Polyacrylonitrile", Handbook of Thermoplastics (CRC Press): 157–186, 2016, ISBN 978-0-429-10162-5
- ↑ 15.0 15.1 David García Cerdá, Antonio Martín Ballester, Alicia Aliena-Valero, Anna Carabén-Redaño, José M. Lloris (August 2015), "Use of cyanoacrylate adhesives in general surgery", Surgery Today 45 (8): 939–956, doi:10.1007/s00595-014-1056-4, PMID 25344231
- ↑ Hans Beyer und Wolfgang Walter: Organische Chemie, S. Hirzel Verlag, Stuttgart, 22. Auflage, 1991, ISBN 3-7776-0485-2, S. 266–269.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitriles". doi:10.1351/goldbook.N04151
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "carbonitriles". doi:10.1351/goldbook.C00838
- ↑ 19.0 19.1 Karl-Heinz Hellwich (1998), Chemische Nomenklatur: die systematische Benennung organisch-chemischer Verbindungen ; ein Lehrbuch für Pharmazie- und Chemiestudenten (3., überarb. Aufl ed.), Eschborn: Govi-Verl, ISBN 3-7741-1095-6
- ↑ Kurt Peter C. Vollhardt, Neil Eric Schore (2011), Organische Chemie. Hauptbd. (5. Aufl ed.), Weinheim: Wiley-VCH, ISBN 978-3-527-32754-6
- ↑ 21.0 21.1 Thomas Laue, Andreas Plagens (1994), "Namen- und Schlagwort-Reaktionen der Organischen Chemie", Teubner Studienbücher Chemie, doi:10.1007/978-3-322-94726-0, ISBN 978-3-519-03526-8
- ↑ K. R. Lynn, Peter E. Yankwich (January 1961), "Cyanide Carbon Isotope Fractionation in the Reaction of Cyanide Ion and Methyl Iodide. Carbon Isotope Effect in the Hydrolysis of Methyl Iodide", Journal of the American Chemical Society 83 (1): 53–57, doi:10.1021/ja01462a010, Bibcode: 1961JAChS..83...53L
- ↑ G. E. Ham, Jane Stevens (December 1962), "Reaction of 1,2-Dihaloethanes with Sodium Cyanide", The Journal of Organic Chemistry 27 (12): 4638–4639, doi:10.1021/jo01059a504
- ↑ Cinzia Chiappe, Daniela Pieraccini, Paola Saullo (2003-08-01), "Nucleophilic Displacement Reactions in Ionic Liquids: Substrate and Solvent Effect in the Reaction of NaN 3 and KCN with Alkyl Halides and Tosylates", The Journal of Organic Chemistry 68 (17): 6710–6715, doi:10.1021/jo026838h, PMID 12919037
- ↑ W. Nagata, M. Yoshioka, S. Hirai (June 1972), "Hydrocyanation. IV. New hydrocyanation methods using hydrogen cyanide and an alkylaluminum, and an alkylaluminum cyanide", Journal of the American Chemical Society 94 (13): 4635–4643, doi:10.1021/ja00768a037, Bibcode: 1972JAChS..94.4635N
- ↑ W. Nagata, M. Yoshioka, T. Okumura (1970), "Hydrocyanation. Part X. Cleavage of epoxides with hydrogen cyanide and triethylaluminium and with diethylaluminium cyanide", Journal of the Chemical Society C: Organic (17): 2365, doi:10.1039/j39700002365
- ↑ Jeffrey C. Mullis, William P. Weber (July 1982), "Regiospecificity of reactions of epoxides and oxetanes with trimethylsilyl cyanide", The Journal of Organic Chemistry 47 (15): 2873–2875, doi:10.1021/jo00136a011
- ↑ Hongru Zhang, Xin Su, Kaiwu Dong (2020), "Recent progress in transition-metal-catalyzed hydrocyanation of nonpolar alkenes and alkynes", Organic & Biomolecular Chemistry 18 (3): 391–399, doi:10.1039/C9OB02374G
- ↑ "ISOBUTYRONITRILE". Organic Syntheses 25: 61. 1945. doi:10.15227/orgsyn.025.0061.
- ↑ "2-ETHYLHEXANONITRILE". Organic Syntheses 32: 65. 1952. doi:10.15227/orgsyn.032.0065.
- ↑ 31.0 31.1 Muthupandian Ganesan, Paramathevar Nagaraaj (2020), "Recent developments in dehydration of primary amides to nitriles", Organic Chemistry Frontiers 7 (22): 3792–3814, doi:10.1039/D0QO00843E
- ↑ 32.0 32.1 Dilip Konwar, Monalisa Boruah, Gautom Kumar Sarmah, Nayan Kamal Bhattacharyya, Naleen Borthakur, Birendra Nath Goswami, Kumar Ranjan Boruah (November 2001), "Aluminium Chloride and Sodium Iodide (AlCl3-NaI): A Versatile Dehydrating Agent", Journal of Chemical Research 2001 (11): 490–492, doi:10.3184/030823401103168604
- ↑ 33.0 33.1 Imen Talbi, Mohamed Lotfi Efrit, Soufiane Touil (2018-05-31), "Efficient New Protocols for Converting Primary Amides into Nitriles Initiated by P(NMe 2 ) 3 , PCl 3 , or P(OPh) 3", ACS Omega 3 (5): 5078–5082, doi:10.1021/acsomega.8b00544, PMID 31458722
- ↑ Muthupandian Ganesan (2021-12-22), "Methods for Direct Conversion of Primary Nitroalkanes to Nitriles", Current Organic Chemistry 25 (24): 2990–3003, doi:10.2174/1385272825666211126124835
- ↑ Z. Shahsavari-Fard, A. R. Sardarian (March 2011), "Diethyl chlorophosphate: A new alternative reagent for dehydration of primary amides to nitriles in solvent and solvent-free conditions", Journal of the Iranian Chemical Society 8 (1): 204–208, doi:10.1007/BF03246217
- ↑ "2-ETHYLHEXANONITRILE", Organic Syntheses 32: 65, 1952, doi:10.15227/orgsyn.032.0065
- ↑ Harry Babad, Andrew G. Zeiler (1973-02-01), "Chemistry of phosgene", Chemical Reviews 73 (1): 75–91, doi:10.1021/cr60281a005, Bibcode: 1973ChRv...73...75B
- ↑ Mohammed H. Al-Huniti, Mitchell P. Croatt (October 2019), "Metal-Catalyzed Dehydration of Primary Amides to Nitriles", Asian Journal of Organic Chemistry 8 (10): 1791–1799, doi:10.1002/ajoc.201900343
- ↑ Hiroyuki Okabe, Asuka Naraoka, Takahiro Isogawa, Shunsuke Oishi, Hiroshi Naka (2019-06-21), "Acceptor-Controlled Transfer Dehydration of Amides to Nitriles", Organic Letters 21 (12): 4767–4770, doi:10.1021/acs.orglett.9b01657, PMID 31184196
- ↑ Stephan Enthaler (September 2011), "Straightforward Iron-Catalyzed Synthesis of Nitriles by Dehydration of Primary Amides", European Journal of Organic Chemistry 2011 (25): 4760–4763, doi:10.1002/ejoc.201100754
- ↑ Stephan Enthaler (2011-08-16), "Straightforward Uranium-Catalyzed Dehydration of Primary Amides to Nitriles", Chemistry: A European Journal 17 (34): 9316–9319, doi:10.1002/chem.201101478, PMID 21728201
- ↑ Stephan Enthaler, Shigeyoshi Inoue (2012-01-02), "An Efficient Zinc-Catalyzed Dehydration of Primary Amides to Nitriles", Chemistry: An Asian Journal 7 (1): 169–175, doi:10.1002/asia.201100493, PMID 21956861
- ↑ Shekharappa, L. Roopesh Kumar, C. Srinivasulu, Vommina V. Sureshbabu (March 2021), "Dehydration of Chiral α-Amides to Chiral α-Nitriles Under the Appel Reaction Conditions", International Journal of Peptide Research and Therapeutics 27 (1): 497–502, doi:10.1007/s10989-020-10101-y
- ↑ Richard S. Monson, Deggary N. Priest (1971-09-01), "Dehydration of Amides to Nitriles Initiated by Hexamethylphosphoric Triamide", Canadian Journal of Chemistry 49 (17): 2897–2898, doi:10.1139/v71-480
- ↑ Krishnappa Manjula, Mohamed Afzal Pasha (May 2007), "Rapid Method of Converting Primary Amides to Nitriles and Nitriles to Primary Amides by ZnCl 2 using Microwaves under Different Reaction Conditions", Synthetic Communications 37 (9): 1545–1550, doi:10.1080/00397910701230147
- ↑ Rui Ding, Yongguo Liu, Mengru Han, Wenyi Jiao, Jiaqi Li, Hongyu Tian, Baoguo Sun (2018-10-19), "Synthesis of Nitriles from Primary Amides or Aldoximes under Conditions of a Catalytic Swern Oxidation", The Journal of Organic Chemistry 83 (20): 12939–12944, doi:10.1021/acs.joc.8b02190, PMID 30240220
- ↑ Kazuaki Ishihara, Yoshiro Furuya, Hisashi Yamamoto (2002-08-16), "Rhenium(VII) Oxo Complexes as Extremely Active Catalysts in the Dehydration of Primary Amides and Aldoximes to Nitriles", Angewandte Chemie 114 (16): 3109, doi:10.1002/1521-3757(20020816)114:16<3109::AID-ANGE3109>3.0.CO;2-K, Bibcode: 2002AngCh.114.3109I
- ↑ Chill, Samuel T.; Mebane, Robert C. (18 September 2009). "A Facile One-Pot Conversion of Aldehydes into Nitriles". Synthetic Communications 39 (20): 3601–3606. doi:10.1080/00397910902788174.
- ↑ C. Fizet; J. Streith (1974). "Hydroxylamine-O-sulfonic acid: A convenient reagent for the oxidative conversion of aldehydes into nitriles" (in German). Tetrahedron Lett. 15 (36): 3187–3188. doi:10.1016/S0040-4039(01)91857-X.
- ↑ Jiban K. Chakrabarti, Terrence M. Hotten (1972), "A new route to nitriles. Dehydration of aldoximes using 2,4,6-trichloro-s-triazine (cyanuric chloride)", Journal of the Chemical Society, Chemical Communications (22): 1226, doi:10.1039/c39720001226
- ↑ "Mild and Efficient Dehydration of Oximes to Nitriles Mediated by the Burgess Reagent", Synlett 2000 (8): 1169–1171, 2000, doi:10.1055/s-2000-6752
- ↑ Ziad Moussa, Saleh A. Ahmed, Ahmad S. ElDouhaibi, Shaya Y. Al-Raqa (April 2010), "NMR Studies and electrophilic properties of triphenylphosphine–trifluoromethanesulfonic anhydride; a remarkable dehydrating reagent system for the conversion of aldoximes into nitriles", Tetrahedron Letters 51 (14): 1826–1831, doi:10.1016/j.tetlet.2010.01.119
- ↑ Kengo Hyodo, Saki Kitagawa, Masayuki Yamazaki, Kingo Uchida (2016-05-06), "Iron-Catalyzed Dehydration of Aldoximes to Nitriles Requiring Neither Other Reagents Nor Nitrile Media", Chemistry: An Asian Journal 11 (9): 1348–1352, doi:10.1002/asia.201600085, PMID 26910510
- ↑ Philipp Rommelmann, Tobias Betke, Harald Gröger (2017-10-20), "Synthesis of Enantiomerically Pure N -Acyl Amino Nitriles via Catalytic Dehydration of Oximes and Application in a de Novo Synthesis of Vildagliptin", Organic Process Research & Development 21 (10): 1521–1527, doi:10.1021/acs.oprd.7b00169
- ↑ 55.0 55.1 Kazuya Yamaguchi, Hiroshi Fujiwara, Yoshiyuki Ogasawara, Miyuki Kotani, Noritaka Mizuno (2007-05-18), "A Tungsten–Tin Mixed Hydroxide as an Efficient Heterogeneous Catalyst for Dehydration of Aldoximes to Nitriles", Angewandte Chemie 119 (21): 3996–3999, doi:10.1002/ange.200605004, Bibcode: 2007AngCh.119.3996Y
- ↑ Dongliang Zhang, Yaping Huang, Erlei Zhang, Rong Yi, Chao Chen, Lei Yu, Qing Xu (2018-02-15), "Pd/Mn Bimetallic Relay Catalysis for Aerobic Aldoxime Dehydration to Nitriles", Advanced Synthesis & Catalysis 360 (4): 784–790, doi:10.1002/adsc.201701154
- ↑ Tobias Betke, Jun Higuchi, Philipp Rommelmann, Keiko Oike, Taiji Nomura, Yasuo Kato, Yasuhisa Asano, Harald Gröger (2018-04-16), "Biocatalytic Synthesis of Nitriles through Dehydration of Aldoximes: The Substrate Scope of Aldoxime Dehydratases", ChemBioChem 19 (8): 768–779, doi:10.1002/cbic.201700571
- ↑ Liyuan Lan, Shuai Huang, Yongguo Liu, Baoguo Sun, Hongyu Tian (July 2020), "Preparation and odor characteristics of nitriles derived from aldehydes", Flavour and Fragrance Journal 35 (4): 425–434, doi:10.1002/ffj.3581
- ↑ Dylan J. Quinn, Graham J. Haun, Gustavo Moura-Letts (August 2016), "Direct synthesis of nitriles from aldehydes with hydroxylamine-O-sulfonic acid in acidic water", Tetrahedron Letters 57 (34): 3844–3847, doi:10.1016/j.tetlet.2016.07.047
- ↑ Xiao-De An, Shouyun Yu (2015-10-16), "Direct Synthesis of Nitriles from Aldehydes Using an O -Benzoyl Hydroxylamine (BHA) as the Nitrogen Source", Organic Letters 17 (20): 5064–5067, doi:10.1021/acs.orglett.5b02547
- ↑ Antonella Leggio, Emilia Lucia Belsito, Sonia Gallo, Angelo Liguori (April 2017), "One-pot conversion of aldehydes to nitriles mediated by TiCl 4", Tetrahedron Letters 58 (15): 1512–1514, doi:10.1016/j.tetlet.2017.03.007
- ↑ "Lewis Acid Reagents Edited by H. Yamamoto. Oxford University Press: Oxford, UK. 1999. 270 pp. £75.00, ISBN 0 19 850099 8.", Organic Process Research & Development 3 (4): 292, 1999-04-15, doi:10.1021/op990022+
- ↑ Jitendra Gurjar, Jorick Bater, Valery V. Fokin (2019-02-06), "Sulfuryl Fluoride Mediated Conversion of Aldehydes to Nitriles", Chemistry: A European Journal 25 (8): 1906–1909, doi:10.1002/chem.201805175, Bibcode: 2019ChEuJ..25.1906G
- ↑ Albert M. van Leusen, Piet G. Oomkes (January 1980), "One-Step Conversion of Aldehydes to Nitriles. Introduction of a One-Carbon Unit", Synthetic Communications 10 (5): 399–403, doi:10.1080/00397918008061830
- ↑ Otto H. Oldenziel, Daan Van Leusen, Albert M. Van Leusen (September 1977), "Chemistry of sulfonylmethyl isocyanides. 13. A general one-step synthesis of nitriles from ketones using tosylmethyl isocyanide. Introduction of a one-carbon unit", The Journal of Organic Chemistry 42 (19): 3114–3118, doi:10.1021/jo00439a002
- ↑ Niamh Disney, Megan Smyth, Scott Wharry, Thomas S. Moody, Marcus Baumann (2024), "A cyanide-free synthesis of nitriles exploiting flow chemistry", Reaction Chemistry & Engineering 9 (2): 349–354, doi:10.1039/D3RE00458A
- ↑ Schümperli, Martin T.; Hammond, Ceri; Hermans, Ive (2021). "Developments in the Aerobic Oxidation of Amines". ACS Catal. 2 (6): 1108–1117. doi:10.1021/cs300212q.
- ↑ Yamazaki, Shigekazu; Yamazaki, Yasuyuki (1990). "Nickel-catalyzed dehydrogenation of amines to nitriles". Bulletin of the Chemical Society of Japan 63 (1): 301–303. doi:10.1246/bcsj.63.301.
- ↑ Chen, Fen-Er; Kuang, Yun-Yan; Hui-Fang, Dai; Lu, Liang (2003). "A Selective and Mild Oxidation of Primary Amines to Nitriles with Trichloroisocyanuric Acid". Synthesis 17 (17): 2629–2631. doi:10.1055/s-2003-42431.
- ↑ Schäfer, H. J.; Feldhues, U. (1982). "Oxidation of Primary Aliphatic Amines to Nitriles at the Nickel Hydroxide Electrode". Synthesis 1982 (2): 145–146. doi:10.1055/s-1982-29721.
- ↑ Xu, Zhining; Kovács, Ervin (2024). "Beyond traditional synthesis: Electrochemical approaches to amine oxidation for nitriles and imines". ACS Org Inorg Au 4 (5): 471–484. doi:10.1021/acsorginorgau.4c00025. PMID 39371318.
- ↑ M. F. Semmelhack, Christopher R. Schmid (October 1983), "Nitroxyl-mediated electro-oxidation of amines to nitriles and carbonyl compounds", Journal of the American Chemical Society 105 (22): 6732–6734, doi:10.1021/ja00360a042, Bibcode: 1983JAChS.105.6732S
- ↑ Kyle M. Lambert, James M. Bobbitt, Sherif A. Eldirany, Liam E. Kissane, Rose K. Sheridan, Zachary D. Stempel, Francis H. Sternberg, William F. Bailey (2016-04-04), "Metal-Free Oxidation of Primary Amines to Nitriles through Coupled Catalytic Cycles", Chemistry: A European Journal 22 (15): 5156–5159, doi:10.1002/chem.201600549, PMID 26868873
- ↑ Yasunari Maeda, Takahiro Nishimura, Sakae Uemura (December 2003), "Copper-Catalyzed Oxidation of Amines with Molecular Oxygen", Bulletin of the Chemical Society of Japan 76 (12): 2399–2403, doi:10.1246/bcsj.76.2399
- ↑ Pollak, Peter; Romeder, Gérard; Hagedorn, Ferdinand; Gelbke, Heinz-Peter (2000). "Ullmann's Encyclopedia of Industrial Chemistry". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a17_363.
- ↑ Christiane Janke, Jörg Radnik, Ursula Bentrup, Andreas Martin, Angelika Brückner (2009-11-30), "Vanadium-Containing Oxynitrides: Effective Catalysts for the Ammoxidation of 3-Picoline", ChemCatChem 1 (4): 485–491, doi:10.1002/cctc.200900180
- ↑ 77.0 77.1 77.2 David Nakles, Richard Luthy, George Wong-Chong (2005-12-09), "Manufacture and the Use of Cyanide", Cyanide in Water and Soil (CRC Press): 41–55
- ↑ Irina P. Beletskaya, Alexander S. Sigeev, Alexander S. Peregudov, Pavel V. Petrovskii (November 2004), "Catalytic Sandmeyer cyanation as a synthetic pathway to aryl nitriles", Journal of Organometallic Chemistry 689 (23): 3810–3812, doi:10.1016/j.jorganchem.2004.07.019
- ↑ C. Frederick Koelsch (August 1936), "Some Applications of the Rosenmund-v. Braun Nitrile Synthesis", Journal of the American Chemical Society 58 (8): 1328–1330, doi:10.1021/ja01299a004, Bibcode: 1936JAChS..58.1328K
- ↑ Naoto Chatani, Terukiyo Hanafusa (November 1986), "Transition-metal-catalyzed reactions of trimethylsilyl cyanide. 4. Palladium-catalyzed cyanation of aryl halides by trimethylsilyl cyanide", The Journal of Organic Chemistry 51 (24): 4714–4716, doi:10.1021/jo00374a041
- ↑ Shunichi Murahashi, Takeshi Naota, Nobuyuki Nakajima (March 1986), "Palladium-catalyzed decarbonylation of acyl cyanides", The Journal of Organic Chemistry 51 (6): 898–901, doi:10.1021/jo00356a029
- ↑ Mark Sundermeier, Alexander Zapf, Matthias Beller, Jürgen Sans (September 2001), "A new palladium catalyst system for the cyanation of aryl chlorides", Tetrahedron Letters 42 (38): 6707–6710, doi:10.1016/S0040-4039(01)01390-9
- ↑ Todd D. Senecal, Wei Shu, Stephen L. Buchwald (2013-09-16), "A General, Practical Palladium-Catalyzed Cyanation of (Hetero)Aryl Chlorides and Bromides", Angewandte Chemie 125 (38): 10219–10223, doi:10.1002/ange.201304188, Bibcode: 2013AngCh.12510219S
- ↑ Florian Glöcklhofer, Markus Lunzer, Johannes Fröhlich (2015-04-01), "Facile Synthesis of Cyanoarenes from Quinones by Reductive Aromatization of Cyanohydrin Intermediates", Synlett 26 (7): 950–952, doi:10.1055/s-0034-1380150
- ↑ Jonathan T. Reeves, Christian A. Malapit, Frederic G. Buono, Kanwar P. Sidhu, Maurice A. Marsini, C. Avery Sader, Keith R. Fandrick, Carl A. Busacca, Chris H. Senanayake (2015-07-29), "Transnitrilation from Dimethylmalononitrile to Aryl Grignard and Lithium Reagents: A Practical Method for Aryl Nitrile Synthesis", Journal of the American Chemical Society 137 (29): 9481–9488, doi:10.1021/jacs.5b06136, PMID 26151426, Bibcode: 2015JAChS.137.9481R
- ↑ "o-Tolunitrile and p-Tolunitrile" H. T. Clarke and R. R. Read Org. Synth. 1941, Coll. Vol. 1, 514.
- ↑ Gregory, Robert J. H. (1999). "Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications". Chemical Reviews 99 (12): 3649–3682. doi:10.1021/cr9902906. PMID 11849033. Bibcode: 1999ChRv...99.3649G.
- ↑ Hiroshi Ohno, Atsunori Mori, Shohei Inoue (February 1993), "Lanthanoid(III) Alkoxides as Novel Catalysts for a Rapid Transhydrocyanation from Acetone Cyanohydrin to Aldehydes and Ketones", Chemistry Letters 22 (2): 375–378, doi:10.1246/cl.1993.375
- ↑ David A. Evans, Gary L. Carroll, Larry K. Truesdale (April 1974), "Synthetic applications of trimethylsilyl cyanide. Efficient synthesis of β-aminomethyl alcohols", The Journal of Organic Chemistry 39 (7): 914–917, doi:10.1021/jo00921a012
- ↑ Shu Kobayashi, Yoshikazu Tsuchiya, Teruaki Mukaiyama (April 1991), "A Facile Synthesis of Cyanohydrin Trimethylsilyl Ethers by the Addition Reaction of Trimethylsilyl Cyanide with Aldehydes under Basic Condition", Chemistry Letters 20 (4): 537–540, doi:10.1246/cl.1991.537
- ↑ Yuri N. Belokon', Susana Caveda-Cepas, Brendan Green, Nicolai S. Ikonnikov, Viktor N. Khrustalev, Vladimir S. Larichev, Margarita A. Moscalenko, Michael North, Charles Orizu, Vitali I. Tararov, Michela Tasinazzo, Galina I. Timofeeva, Lidia V. Yashkina (1999-04-01), "The Asymmetric Addition of Trimethylsilyl Cyanide to Aldehydes Catalyzed by Chiral (Salen)Titanium Complexes", Journal of the American Chemical Society 121 (16): 3968–3973, doi:10.1021/ja984197v, Bibcode: 1999JAChS.121.3968B
- ↑ 92.0 92.1 Nobuhito Kurono, Takeshi Ohkuma (2016-02-05), "Catalytic Asymmetric Cyanation Reactions", ACS Catalysis 6 (2): 989–1023, doi:10.1021/acscatal.5b02184
- ↑ Siegfried Hünig, Rainer Schaller (January 1982), "The Chemistry of Acyl Cyanides", Angewandte Chemie International Edition in English 21 (1): 36–49, doi:10.1002/anie.198200361
- ↑ Harald Gröger, Yasuhisa Asano (2020), "Cyanide-Free Enantioselective Catalytic Strategies for the Synthesis of Chiral Nitriles", The Journal of Organic Chemistry 85 (10): 6243–6251, doi:10.1021/acs.joc.9b02773, PMID 32250626
- ↑ W. Nagata and M. Yoshioka (1988). "Diethylaluminum cyanide". Organic Syntheses. http://www.orgsyn.org/demo.aspx?prep=cv6p0436.; Collective Volume, 6, pp. 436
- ↑ W. Nagata, M. Yoshioka, and M. Murakami (1988). "Preparation of cyano compounds using alkylaluminum intermediates: 1-cyano-6-methoxy-3,4-dihydronaphthalene". Organic Syntheses. http://www.orgsyn.org/demo.aspx?prep=cv6p0307.; Collective Volume, 6, pp. 307
- ↑ Reynold C. Fuson; Oscar R. Kreimeier; Gilbert L. Nimmo (1930). "Ring Closures in the Cyclobutane Series. II. Cyclization Of α,α′-Dibromo-Adipic Esters". J. Am. Chem. Soc. 52 (10): 4074–4076. doi:10.1021/ja01373a046.
- ↑ J. Houben, Walter Fischer (1930) "Über eine neue Methode zur Darstellung cyclischer Nitrile durch katalytischen Abbau (I. Mitteil.)," Berichte der deutschen chemischen Gesellschaft (A and B Series) 63 (9): 2464 – 2472. doi:10.1002/cber.19300630920
- ↑ Hiegel, Gene; Lewis, Justin; Bae, Jason (2004). "Conversion of α-Amino Acids into Nitriles by Oxidative Decarboxylation with Trichloroisocyanuric Acid". Synthetic Communications 34 (19): 3449–3453. doi:10.1081/SCC-200030958.
- ↑ Hampson, N; Lee, J; MacDonald, K (1972). "The oxidation of amino compounds at anodic silver". Electrochimica Acta 17 (5): 921–955. doi:10.1016/0013-4686(72)90014-X.
- ↑ Dakin, Henry Drysdale (1916). "The Oxidation of Amino-Acids to Cyanides". Biochemical Journal 10 (2): 319–323. doi:10.1042/bj0100319. PMID 16742643.
- ↑ Yoshiaki Nakao, Tamejiro Hiyama (2008-01-01), "Nickel-catalyzed carbocyanation of alkynes", Pure and Applied Chemistry 80 (5): 1097–1107, doi:10.1351/pac200880051097
- ↑ Haitham Hassan, Vincent Pirenne, Maren Wissing, Chahinaz Khiar, Ashique Hussain, Frédéric Robert, Yannick Landais (2017-04-03), "Free-Radical Carbocyanation of Olefins", Chemistry: A European Journal 23 (19): 4651–4658, doi:10.1002/chem.201605946, PMID 28094885
- ↑ Laurent Vanoye, Ahmad Hammoud, Hélène Gérard, Alexandra Barnes, Régis Philippe, Pascal Fongarland, Claude de Bellefon, Alain Favre-Réguillon (2019-11-01), "Direct Synthesis of Nitriles from Carboxylic Acids Using Indium-Catalyzed Transnitrilation: Mechanistic and Kinetic Study", ACS Catalysis 9 (11): 9705–9714, doi:10.1021/acscatal.9b02779
- ↑ Tetsuto Tsunoda, Kaori Uemoto, Chisato Nagino, Megumi Kawamura, Hiroto Kaku, Shô Itô (October 1999), "A facile one-pot cyanation of primary and secondary alcohols. Application of some new Mitsunobu reagents", Tetrahedron Letters 40 (41): 7355–7358, doi:10.1016/S0040-4039(99)01509-9
- ↑ Walter-Georg Veeck, Manfred Regitz (1995), "Triple-bonded Heteroatom Derivatives Other Than Nitriles with Another Heteroatom Attached to the sp-Carbon Atom", Comprehensive Organic Functional Group Transformations (Elsevier): 1151–1160
- ↑ B.A. Phillips, G. Fodor, J. Gal, F. Letourneau, J.J. Ryan (January 1973), "Mechanism of the von Braun amide degradations with carbonyl bromide or phosphorus pentabromide", Tetrahedron 29 (21): 3309–3327, doi:10.1016/S0040-4020(01)93483-0
- ↑ Kukushkin, V. Yu.; Pombeiro, A. J. L. (2005). "Metal-mediated and metal-catalyzed hydrolysis of nitriles". Inorg. Chim. Acta 358: 1–21. doi:10.1016/j.ica.2004.04.029.
- ↑ Abbas, Khamis A. (2008-01-01). "Substituent Effects on the Hydrolysis of p-Substituted Benzonitriles in Sulfuric Acid Solutions at (25.0± 0.1) °C". Zeitschrift für Naturforschung A 63 (9): 603–608. doi:10.1515/zna-2008-0912. ISSN 1865-7109. Bibcode: 2008ZNatA..63..603A.
- ↑ Dahye Kang; Jinwoo Lee; Hee-Yoon Lee (2012). "Anhydrous Hydration of Nitriles to Amides: p-Carbomethoxybenzamide". Organic Syntheses 89: 66. doi:10.15227/orgsyn.089.0066.
- ↑ Barrault, J.; Pouilloux, Y. (1997). "Catalytic Amination Reactions: Synthesis of fatty amines. Selectivity control in presence of multifunctional catalysts". Catalysis Today 1997 (2): 137–153. doi:10.1016/S0920-5861(97)00006-0.
- ↑ Arseniyadis, Siméon; Kyler, Keith S.; Watt, David S. (1984). "Addition and Substitution Reactions of Nitrile-Stabilized Carbanions". Organic Reactions. pp. 1–364. doi:10.1002/0471264180.or031.01. ISBN 978-0-471-26418-7.
- ↑ Yang, Xun; Fleming, Fraser F. (2017). "C- and N-Metalated Nitriles: The Relationship between Structure and Selectivity". Accounts of Chemical Research 50 (10): 2556–2568. doi:10.1021/acs.accounts.7b00329. PMID 28930437.
- ↑ Smith, Andri L.; Tan, Paula (2006). "Creatine Synthesis: An Undergraduate Organic Chemistry Laboratory Experiment". J. Chem. Educ. 83 (11): 1654. doi:10.1021/ed083p1654. Bibcode: 2006JChEd..83.1654S.
- ↑ "Moureau-Mignonac Ketimine Synthesis" (in en). Comprehensive Organic Name Reactions and Reagents. Hoboken, NJ, USA: John Wiley & Sons, Inc.. 2010-09-15. pp. 1988–1990. doi:10.1002/9780470638859.conrr446. ISBN 978-0-470-63885-9.
- ↑ 116.0 116.1 The reductive decyanation reaction: chemical methods and synthetic applications Jean-Marc Mattalia, Caroline Marchi-Delapierre, Hassan Hazimeh, and Michel Chanon Arkivoc (AL-1755FR) pp. 90–118 2006 Article
- ↑ Berkoff, Charles E.; Rivard, Donald E.; Kirkpatrick, David; Ives, Jeffrey L. (1980). "The Reductive Decyanation of Nitriles by Alkali Fusion". Synthetic Communications 10 (12): 939–945. doi:10.1080/00397918008061855.
- ↑ Franchimont, Antoine Paul Nicholas (1872). "Ueber die Dibenzyldicarbonsäure". Berichte der Deutschen Chemischen Gesellschaft 5 (2): 1048–1050. doi:10.1002/cber.187200502138. https://babel.hathitrust.org/cgi/pt?id=uc1.b3481750;seq=1012.
- ↑ Yoshiaki Nakao; Akira Yada; Shiro Ebata; Tamejiro Hiyama (2007). "A Dramatic Effect of Lewis-Acid Catalysts on Nickel-Catalyzed Carbocyanation of Alkynes". J. Am. Chem. Soc. 129 (9): 2428–2429. doi:10.1021/ja067364x. PMID 17295484. Bibcode: 2007JAChS.129.2428N.
- ↑ Rach, S. F.; Kühn, F. E. (2009). "Nitrile Ligated Transition Metal Complexes with Weakly Coordinating Counteranions and Their Catalytic Applications". Chemical Reviews 109 (5): 2061–2080. doi:10.1021/cr800270h. PMID 19326858.
- ↑ Moss, G. P.; Smith, P. A. S.; Tavernier, D. (1995-01-01). "Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995)". Pure and Applied Chemistry 67 (8–9): 1307–1375. doi:10.1351/pac199567081307. Bibcode: 1995PApCh..67.1307M.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "cyanates". doi:10.1351/goldbook.C01485
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "thiocyanates". doi:10.1351/goldbook.T06353
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "selenocyanates". doi:10.1351/goldbook.S05573
- ↑ Prabhath, M.; Williams, Luke; Bhat, Shreesha; Sharma, Pallavi (2017-04-12). "Recent Advances in Cyanamide Chemistry: Synthesis and Applications". Molecules 22 (4): 615. doi:10.3390/molecules22040615. PMID 28417938.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrile oxides". doi:10.1351/goldbook.N04150
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrile sulfides". doi:10.1351/goldbook.N04152
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrile imides". doi:10.1351/goldbook.N04148
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrilium ions". doi:10.1351/goldbook.N04156
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrile ylides". doi:10.1351/goldbook.N04153
- ↑ Template:March4th
- ↑ 132.0 132.1 132.2 Smith, Michael B.; March, Jerry (2007). March's Advanced Organic Chemistry (6th ed.). John Wiley & Sons. ISBN 978-0-471-72091-1.
- ↑ Grundmann, Ch. (1970). "Nitrile oxides". in Rappoport, Zvi. The Chemistry of the Cyano Group. PATai's Chemistry of Functional Groups. p. 794. doi:10.1002/9780470771242.ch14. ISBN 978-0-471-70913-8.
- ↑ 134.0 134.1 Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2001). Organic Chemistry (1st ed.). Oxford University Press. ISBN 978-0-19-850346-0.
- ↑ Hagedorn, Alfred A.; Miller, Bryan J.; Nagy, Jon O. (1980). "Direct synthesis of the antitumor agent erythro-α‑amino-3‑bromo-4,5‑dihydrooxazole-5‑acetic acid". Tetrahedron Letters (Great Britain: Pergamon) 21: 229–230. doi:10.1016/S0040-4039(00)71175-0none, alluding to a large-scale modification later detailed in Vyas, D. M.; Chiang, Y.; Doyle, T. W. (1984). "A short, efficient total synthesis of (±) acivicin and (±) bromo-acivicin". Tetrahedron Letters (Great Britain: Pergamon) 25 (5): fn. 10. doi:10.1016/S0040-4039(00)99918-0.
- ↑ Argyropoulos, Nikolaos G. (1996). "1,4-Oxa/thia-2-azoles". Comprehensive Heterocyclic Chemistry. 4: Five-membered rings with more than two heteroatoms and fused carbocyclic derivatives. pp. 506–507. doi:10.1016/B978-008096518-5.00092-7. ISBN 978-0-08-096518-5.
- ↑ 137.0 137.1 137.2 137.3 Fraser F. Fleming, Fraser F. Fleming (1999), "Nitrile-containing natural products", Natural Product Reports 16 (5): 597–606, doi:10.1039/a804370a
- ↑ 138.0 138.1 138.2 138.3 Camille Scotti, James W. Barlow (May 2022), "Natural Products Containing the Nitrile Functional Group and Their Biological Activities", Natural Product Communications 17 (5): 1934578X2210999, doi:10.1177/1934578X221099973
- ↑ Anete C Ferraz, Miriam Elizabeth M Angelucci, Mariana L Da Costa, Ilza R Batista, Bras H De Oliveira, Claudio Da Cunha (July 1999), "Pharmacological Evaluation of Ricinine, a Central Nervous System Stimulant Isolated from Ricinus communis", Pharmacology Biochemistry and Behavior 63 (3): 367–375, doi:10.1016/S0091-3057(99)00007-6, PMID 10418776
- ↑ R. Mukherjee, A. Chatterjee (January 1966), "Structure and synthesis of nudiflorine", Tetrahedron 22 (4): 1461–1466, doi:10.1016/S0040-4020(01)99443-8
- ↑ M.Soledade C Pedras, Corwin M Nycholat, Sabine Montaut, Yiming Xu, Abdul Q Khan (March 2002), "Chemical defenses of crucifers: elicitation and metabolism of phytoalexins and indole-3-acetonitrile in brown mustard and turnip", Phytochemistry 59 (6): 611–625, doi:10.1016/S0031-9422(02)00026-2, PMID 11867093, Bibcode: 2002PChem..59..611P
- ↑ A. Bellirou, A. Bouali, B. Bouammali, N. Boukhatem, B.N. Elmtili, A. Hamal, M. El-Mourabit (March 2005), "Extraction of simmondsin and oil in one step from jojoba seeds", Industrial Crops and Products 21 (2): 229–233, doi:10.1016/j.indcrop.2004.04.007
- ↑ Adolf Nahrstedt, Victor Wray (1990), "Structural revision of a putative cyanogenic glucoside from Ilex aquifolium", Phytochemistry 29 (12): 3934–3936, doi:10.1016/0031-9422(90)85364-L, Bibcode: 1990PChem..29.3934N
- ↑ Wendy K. Swenson, John E. Dunn, Eric E. Conn (January 1987), "Cyanogenesis in Acacia sutherlandii", Phytochemistry 26 (6): 1835–1836, doi:10.1016/S0031-9422(00)82299-2, Bibcode: 1987PChem..26.1835S
- ↑ Martin G. Ettlinger, Jerzy W. Jaroszewski, Søren Rosendal Jensen, Bent Juhl Nielsen, Frederick Nartey (1977), "Proacacipetalin and acacipetalin", Journal of the Chemical Society, Chemical Communications (24): 952, doi:10.1039/c39770000952
- ↑ K Shanker, M Gupta, S Srivastava, D Bawankule, A Pal, S Khanuja (2007), "Determination of bioactive nitrile glycoside(s) in drumstick (Moringa oleifera) by reverse phase HPLC", Food Chemistry 105 (1): 376–382, doi:10.1016/j.foodchem.2006.12.034
- ↑ F. W. Stamler (1955-10-01), "Reproduction in Rats Fed Lathyrus Peas or Aminonitriles", Experimental Biology and Medicine 90 (1): 294–298, doi:10.3181/00379727-90-22013, PMID 13273428
- ↑ E. D. Schilling, F. M. Strong (May 1954), "ISOLATION, STRUCTURE AND SYNTHESIS OF a LATHYRUS FACTOR FROM L. ODORATUS1", Journal of the American Chemical Society 76 (10): 2848, doi:10.1021/ja01639a084, Bibcode: 1954JAChS..76Q2848S
- ↑ Mohammadali Torbati, Hossein Nazemiyeh, Farzaneh Lotfipour, Solmaz Asnaashari, Mahboob Nemati, Fatemeh Fathiazad (2013), "Composition and Antibacterial Activity of Heracleum Transcaucasicum and Heracleum Anisactis Aerial Parts Essential Oil", Advanced Pharmaceutical Bulletin 3 (2): 415–418, doi:10.5681/APB.2013.066, PMID 24312869
- ↑ Peter Lorenz, Sarina Duckstein, Jürgen Conrad, Matthias Knödler, Ulrich Meyer, Florian C. Stintzing (February 2012), "An Approach to the Chemotaxonomic Differentiation of Two European Dog's Mercury Species: Mercurialis annua L. and M. perennis L.", Chemistry & Biodiversity 9 (2): 282–297, doi:10.1002/cbdv.201100341
- ↑ K.L. Mikolajczak (January 1977), "Cyanolipids", Progress in the Chemistry of Fats and Other Lipids 15 (2): 97–130, doi:10.1016/0079-6832(77)90013-1, PMID 327513
- ↑ David S. Seigler, Wanda Kawahara (January 1976), "New reports of cyanolipids from sapindaceous plants", Biochemical Systematics and Ecology 4 (4): 263–265, doi:10.1016/0305-1978(76)90050-8, Bibcode: 1976BioSE...4..263S
- ↑ E E Conn (June 1980), "Cyanogenic Compounds", Annual Review of Plant Physiology 31 (1): 433–451, doi:10.1146/annurev.pp.31.060180.002245
- ↑ 154.0 154.1 Pablo Díaz-Rueda, Laura Morales de los Ríos, Luis C Romero, Irene García (2023-10-13), "Old poisons, new signaling molecules: the case of hydrogen cyanide", Journal of Experimental Botany 74 (19): 6040–6051, doi:10.1093/jxb/erad317, PMID 37586035
- ↑ Hieng-Ming Ting, Boon Huat Cheah, Yu-Cheng Chen, Pei-Min Yeh, Chiu-Ping Cheng, Freddy Kuok San Yeo, Ane Kjersti Vie, Jens Rohloff, Per Winge, Atle M. Bones, Ralph Kissen (2020-03-10), "The Role of a Glucosinolate-Derived Nitrile in Plant Immune Responses", Frontiers in Plant Science 11, doi:10.3389/fpls.2020.00257, PMID 32211010, Bibcode: 2020FrPS...11..257T
- ↑ Adam M. Wentzell, Daniel J. Kliebenstein (2008-04-28), "Genotype, Age, Tissue, and Environment Regulate the Structural Outcome of Glucosinolate Activation", Plant Physiology 147 (1): 415–428, doi:10.1104/pp.107.115279, PMID 18359845, Bibcode: 2008PlanP.147..415W
- ↑ Hideji Tanii (March 2017), "Allyl nitrile: Toxicity and health effects", Journal of Occupational Health 59 (2): 104–111, doi:10.1539/joh.16-0147-RA, PMID 28132970
- ↑ Jinghua Yang, Zhangping Li, Jinmin Lian, Guoning Qi, Pibiao Shi, Jiawei He, Zhongyuan Hu, Mingfang Zhang (2020-12-16), "Brassicaceae transcriptomes reveal convergent evolution of super-accumulation of sinigrin", Communications Biology 3 (1), doi:10.1038/s42003-020-01523-x, PMID 33328568
- ↑ David J. Williams, Christa Critchley, Sharon Pun, Mridusmita Chaliha, Timothy J. O'Hare (July 2009), "Differing mechanisms of simple nitrile formation on glucosinolate degradation in Lepidium sativum and Nasturtium officinale seeds", Phytochemistry 70 (11–12): 1401–1409, doi:10.1016/j.phytochem.2009.07.035, PMID 19747700, Bibcode: 2009PChem..70.1401W
- ↑ Ani Radonić, Ivica Blažević, Josip Mastelić, Marina Zekić, Mirjana Skočibušić, Ana Maravić (June 2011), "Phytochemical Analysis and Antimicrobial Activity of Cardaria draba (L.) Desv . Volatiles", Chemistry & Biodiversity 8 (6): 1170–1181, doi:10.1002/cbdv.201000370, PMID 21674789
- ↑ Islamiyat F. Bolarinwa, Caroline Orfila, Michael R.A. Morgan (June 2014), "Amygdalin content of seeds, kernels and food products commercially-available in the UK", Food Chemistry 152: 133–139, doi:10.1016/j.foodchem.2013.11.002, PMID 24444917, Bibcode: 2014FoodC.152..133B
- ↑ C.J Graham (May 2002), "Nonstructural carbohydrate and prunasin composition of peach seedlings fertilized with different nitrogen sources and aluminum", Scientia Horticulturae 94 (1–2): 21–32, doi:10.1016/S0304-4238(01)00345-4, Bibcode: 2002ScHor..94...21G
- ↑ C.J Graham (May 2002), "Nonstructural carbohydrate and prunasin composition of peach seedlings fertilized with different nitrogen sources and aluminum", Scientia Horticulturae 94 (1–2): 21–32, doi:10.1016/S0304-4238(01)00345-4, Bibcode: 2002ScHor..94...21G
- ↑ Jandirk Sendker, Therese Ellendorff, Aljoscha Hölzenbein (2016-07-22), "Occurrence of Benzoic Acid Esters as Putative Catabolites of Prunasin in Senescent Leaves of Prunus laurocerasus", Journal of Natural Products 79 (7): 1724–1729, doi:10.1021/acs.jnatprod.5b01090, PMID 27331617, Bibcode: 2016JNAtP..79.1724S
- ↑ David Chassagne, Jean C. Crouzet, Claude L. Bayonove, Raymond L. Baumes (1996-01-01), "Identification and Quantification of Passion Fruit Cyanogenic Glycosides", Journal of Agricultural and Food Chemistry 44 (12): 3817–3820, doi:10.1021/jf960381t, Bibcode: 1996JAFC...44.3817C
- ↑ David S. Seigler, Guido F. Pauli, Adolf Nahrstedt, Rosemary Leen (August 2002), "Cyanogenic allosides and glucosides from Passiflora edulis and Carica papaya", Phytochemistry 60 (8): 873–882, doi:10.1016/S0031-9422(02)00170-X, PMID 12150815, Bibcode: 2002PChem..60..873S
- ↑ Mateja Senica, Franci Stampar, Robert Veberic, Maja Mikulic-Petkovsek (June 2017), "The higher the better? Differences in phenolics and cyanogenic glycosides in Sambucus nigra leaves, flowers and berries from different altitudes", Journal of the Science of Food and Agriculture 97 (8): 2623–2632, doi:10.1002/jsfa.8085, PMID 27734518, Bibcode: 2017JSFA...97.2623S
- ↑ Rex A. Buhrmester, John E. Ebinger, David S. Seigler (August 2000), "Sambunigrin and cyanogenic variability in populations of Sambucus canadensis L. (Caprifoliaceae)", Biochemical Systematics and Ecology 28 (7): 689–695, doi:10.1016/S0305-1978(99)00105-2, PMID 10854744, Bibcode: 2000BioSE..28..689B
- ↑ Nhat Hao Tran Le, Karl Egil Malterud, Drissa Diallo, Berit Smestad Paulsen, Cecilie Sogn Nergård, Helle Wangensteen (February 2012), "Bioactive polyphenols in Ximenia americana and the traditional use among Malian healers", Journal of Ethnopharmacology 139 (3): 858–862, doi:10.1016/j.jep.2011.12.031, PMID 22212502
- ↑ H. Kofod, R. Eyjólfsson (August 1969), "Cyanogenesis in species of the fern genera Cystopteris and Davalla", Phytochemistry 8 (8): 1509–1511, doi:10.1016/S0031-9422(00)85922-1, Bibcode: 1969PChem...8.1509K
- ↑ Gilles F. Nicollier, Daniel F. Pope, Alonzo C. Thompson (July 1983), "Biological activity of dhurrin and other compounds from Johnson grass (Sorghum halepense)", Journal of Agricultural and Food Chemistry 31 (4): 744–748, doi:10.1021/jf00118a016, Bibcode: 1983JAFC...31..744N
- ↑ Tomas Laursen, Jonas Borch, Camilla Knudsen, Krutika Bavishi, Federico Torta, Helle Juel Martens, Daniele Silvestro, Nikos S. Hatzakis, Markus R. Wenk, Timothy R. Dafforn, Carl Erik Olsen, Mohammed Saddik Motawia, Björn Hamberger, Birger Lindberg Møller, Jean-Etienne Bassard (2016-11-18), "Characterization of a dynamic metabolon producing the defense compound dhurrin in sorghum", Science 354 (6314): 890–893, doi:10.1126/science.aag2347, PMID 27856908, Bibcode: 2016Sci...354..890L, https://research.birmingham.ac.uk/en/publications/90ddc888-b558-4034-a64e-4984a6708865
- ↑ G.W. Butler (February 1965), "The distribution of the cyanoglucosides linamarin and lotaustralin in higher plants", Phytochemistry 4 (1): 127–131, doi:10.1016/S0031-9422(00)86154-3, Bibcode: 1965PChem...4..127B
- ↑ M. P. Cereda, M.C.Y. Mattos (1996), "Linamarin: The Toxic Compound of Cassava", Journal of Venomous Animals and Toxins 2 (1): 06–12, doi:10.1590/S0104-79301996000100002
- ↑ Matthias Onyebuchi Agbo, Daowan Lai, Festus B.C. Okoye, Patience O. Osadebe, Peter Proksch (April 2013), "Antioxidative polyphenols from Nigerian mistletoe Loranthus micranthus (Linn.) parasitizing on Hevea brasiliensis", Fitoterapia 86: 78–83, doi:10.1016/j.fitote.2013.02.006, PMID 23422225
- ↑ R. Lieberei, D. Selmar, B. Biehl (1985), "Metabolization of cyanogenic glucosides in Hevea brasiliensis", Plant Systematics and Evolution 150 (1–2): 49–63, doi:10.1007/BF00985567, Bibcode: 1985PSyEv.150...49L
- ↑ 177.0 177.1 Mika Zagrobelny, Érika de Castro, Birger Møller, Søren Bak (2018-05-03), "Cyanogenesis in Arthropods: From Chemical Warfare to Nuptial Gifts", Insects 9 (2): 51, doi:10.3390/insects9020051, PMID 29751568
- ↑ Ritsuo Nishida, Miriam Rothschild, Rosemary Mummery (May 1994), "Acyanoglucoside, sarmentosin, from the magpie moth, Abraxas grossulariata, geometridae: Lepidoptera", Phytochemistry 36 (1): 37–38, doi:10.1016/S0031-9422(00)97007-9, Bibcode: 1994PChem..36...37N
- ↑ Nanna Bjarnholt, Mirosław Nakonieczny, Andrzej Kędziorski, Diane M. Debinski, Stephen F. Matter, Carl Erik Olsen, Mika Zagrobelny (May 2012), "Occurrence of Sarmentosin and Other Hydroxynitrile Glucosides in Parnassius (Papilionidae) Butterflies and Their Food Plants", Journal of Chemical Ecology 38 (5): 525–537, doi:10.1007/s10886-012-0114-x, PMID 22527055, Bibcode: 2012JCEco..38..525B
- ↑ J. R. Aldrich, S. P. Carroll, W. R. Lusby, M. J. Thompson, J. P. Kochansky, R. M. Waters (January 1990), "Sapindaceae, cyanolipids, and bugs", Journal of Chemical Ecology 16 (1): 199–210, doi:10.1007/BF01021279, PMID 24264907, Bibcode: 1990JCEco..16..199A
- ↑ J.C. Braekman, D. Daloze, J.M. Pasteels (December 1982), "Cyanogenic and other glucosides in a neo-guinean bug Leptocoris isolata: Possible precursors in its host-plant", Biochemical Systematics and Ecology 10 (4): 355–364, doi:10.1016/0305-1978(82)90010-2, Bibcode: 1982BioSE..10..355B
- ↑ Mika Zagrobelny, Karsten Scheibye-Alsing, Niels Bjerg Jensen, Birger Lindberg Møller, Jan Gorodkin, Søren Bak (December 2009), "454 pyrosequencing based transcriptome analysis of Zygaena filipendulae with focus on genes involved in biosynthesis of cyanogenic glucosides", BMC Genomics 10 (1), doi:10.1186/1471-2164-10-574, PMID 19954531, Bibcode: 2009BMCG...10..574Z
- ↑ Adolf Nahrstedt (1993), "Cyanogenesis and foodplants", Phytochemistry and Agriculture (Oxford University Press Oxford): 107–129, ISBN 0-19-857762-1
- ↑ Ljubodrag V. Vujisić, Ivan M. Vučković, Slobodan E. Makarov, Bojan S. Ilić, Dragan Ž. Antić, Milka B. Jadranin, Nina M. Todorović, Ivan V. Mrkić, Vlatka E. Vajs, Luka R. Lučić, Božidar P. M. Ćurčić, Bojan M. Mitić (September 2013), "Chemistry of the sternal gland secretion of the Mediterranean centipede Himantarium gabrielis (Linnaeus, 1767) (Chilopoda: Geophilomorpha: Himantariidae)", Naturwissenschaften 100 (9): 861–870, doi:10.1007/s00114-013-1086-6, PMID 23907296, Bibcode: 2013NW....100..861V
- ↑ Karsten Seidelmann, Heike Weinert, Hans-Jörg Ferenz (December 2003), "Wings and legs are production sites for the desert locust courtship-inhibition pheromone, phenylacetonitrile", Journal of Insect Physiology 49 (12): 1125–1133, doi:10.1016/j.jinsphys.2003.08.005, PMID 14624884, Bibcode: 2003JInsP..49.1125S
- ↑ S. S. Duffey, M. S. Blum, H. M. Fales, S. L. Evans, R. W. Roncadori, D. L. Tiemann, Y. Nakagawa (1977), "Benzoyl cyanide and mandelonitrile benzoate in the defensive secretions of millipedes", Journal of Chemical Ecology 3 (1): 101–113, doi:10.1007/BF00988137, Bibcode: 1977JCEco...3..101D
- ↑ Adrian Brückner, Günther Raspotnig, Katja Wehner, Reinhard Meusinger, Roy A. Norton, Michael Heethoff (2017-03-28), "Storage and release of hydrogen cyanide in a chelicerate ( Oribatula tibialis )", Proceedings of the National Academy of Sciences 114 (13): 3469–3472, doi:10.1073/pnas.1618327114, PMID 28289203, Bibcode: 2017PNAS..114.3469B
- ↑ Guido Cimino, Margherita Gavagnin, Guido Sodano, Aldo Spinella, Giuseppe Strazzullo, Francis J. Schmitz, Gopichand Yalamanchili (May 1987), "Revised structure of bursatellin", The Journal of Organic Chemistry 52 (11): 2301–2303, doi:10.1021/jo00387a037
- ↑ Annika Fagerholm, Damien Habrant, Ari M. Koskinen (2010-01-21), "Calyculins and Related Marine Natural Products as Serine-Threonine Protein Phosphatase PP1 and PP2A Inhibitors and Total Syntheses of Calyculin A, B, and C", Marine Drugs 8 (1): 122–172, doi:10.3390/md80100122, PMID 20161975
- ↑ Samuele Sala, Jane Fromont, Oliver Gomez, Daniel Vuong, Ernest Lacey, Gavin R. Flematti (2019-12-27), "Albanitriles A–G: Antiprotozoal Polyacetylene Nitriles from a Mycale Marine Sponge", Journal of Natural Products 82 (12): 3450–3455, doi:10.1021/acs.jnatprod.9b00840, PMID 31833368, Bibcode: 2019JNAtP..82.3450S
- ↑ C J Knowles (September 1976), "Microorganisms and cyanide", Bacteriological Reviews 40 (3): 652–680, doi:10.1128/br.40.3.652-680.1976, PMID 791236
- ↑ Hideyuki Takahashi, Koohei Nozawa, Ken-ichi Kawai (1996), "Isolation and Structures of Dicyanide Derivatives, Epurpurins A to C, from Emericella purpurea.", Chemical and Pharmaceutical Bulletin 44 (12): 2227–2230, doi:10.1248/cpb.44.2227
- ↑ Marjorie Anchel (November 1958), "Metabolic products of Clitocybe diatreta. I. Diatretyne amide and diatretyne nitrile", Archives of Biochemistry and Biophysics 78 (1): 100–110, doi:10.1016/0003-9861(58)90318-7, PMID 13595907
- ↑ N. G. Heatley, J. S. Stephenson (May 1957), "Identity of 'Nudic Acid B' and 'Diatretyne II'", Nature 179 (4569): 1078, doi:10.1038/1791078a0, Bibcode: 1957Natur.179Q1078H
- ↑ Jan Caspar, Peter Spiteller (2015-03-02), "A Free Cyanohydrin as Arms and Armour of Marasmius oreades", ChemBioChem 16 (4): 570–573, doi:10.1002/cbic.201402453, PMID 25630401
- ↑ Anju Sehrawat, Satyavir S. Sindhu, Bernard R. Glick (February 2022), "Hydrogen cyanide production by soil bacteria: Biological control of pests and promotion of plant growth in sustainable agriculture", Pedosphere 32 (1): 15–38, doi:10.1016/S1002-0160(21)60058-9, Bibcode: 2022Pedos..32...15S
- ↑ Diogo Montes Vidal, Anna-Lena von Rymon-Lipinski, Srinivasa Ravella, Ulrike Groenhagen, Jennifer Herrmann, Nestor Zaburannyi, Paulo H. G. Zarbin, Adithi R. Varadarajan, Christian H. Ahrens, Laure Weisskopf, Rolf Müller, Stefan Schulz (2017-04-03), "Long-Chain Alkyl Cyanides: Unprecedented Volatile Compounds Released by Pseudomonas and Micromonospora Bacteria", Angewandte Chemie International Edition 56 (15): 4342–4346, doi:10.1002/anie.201611940, PMID 28276609
- ↑ Joel P. Cioni, James R. Doroghazi, Kou-San Ju, Xiaomin Yu, Bradley S. Evans, Jaeheon Lee, William W. Metcalf (2014-02-28), "Cyanohydrin Phosphonate Natural Product from Streptomyces regensis", Journal of Natural Products 77 (2): 243–249, doi:10.1021/np400722m, PMID 24437999, Bibcode: 2014JNAtP..77..243C
- ↑ Sanjoy Adak, April L. Lukowski, Rebecca J. B. Schäfer, Bradley S. Moore (2022-02-23), "From Tryptophan to Toxin: Nature's Convergent Biosynthetic Strategy to Aetokthonotoxin", Journal of the American Chemical Society 144 (7): 2861–2866, doi:10.1021/jacs.1c12778, PMID 35142504, Bibcode: 2022JAChS.144.2861A
- ↑ 200.0 200.1 Max P. Bernstein, Samantha F. M. Ashbourn, Scott A. Sandford, Louis J. Allamandola (2004-01-20), "The Lifetimes of Nitriles (CN) and Acids (COOH) during Ultraviolet Photolysis and Their Survival in Space", The Astrophysical Journal 601 (1): 365–370, doi:10.1086/380306, Bibcode: 2004ApJ...601..365B
- ↑ S Green (October 1981), "Interstellar Chemistry: Exotic Molecules in Space", Annual Review of Physical Chemistry 32 (1): 103–138, doi:10.1146/annurev.pc.32.100181.000535, Bibcode: 1981ARPC...32..103G
- ↑ Richard Carter, Mary M. Nijhout (1977-01-28), "Control of Gamete Formation (Exflagellation) in Malaria Parasites", Science 195 (4276): 407–409, doi:10.1126/science.12566, PMID 12566, Bibcode: 1977Sci...195..407C
- ↑ Jean-Claude Guillemin, Miloud Bouyahyi, El Hassan Riague (January 2004), "Prebiotic, planetary and interstellar chemistry starting from compounds detected in the interstellar medium", Advances in Space Research 33 (1): 81–87, doi:10.1016/j.asr.2003.07.015, Bibcode: 2004AdSpR..33...81G
- ↑ 204.0 204.1 204.2 Nicholas F. Wogan, David C. Catling, Kevin J. Zahnle, Roxana Lupu (2023-09-01), "Origin-of-life Molecules in the Atmosphere after Big Impacts on the Early Earth", The Planetary Science Journal 4 (9): 169, doi:10.3847/PSJ/aced83, Bibcode: 2023PSJ.....4..169W
- ↑ 205.0 205.1 Yannick Vallee, Ibrahim Shalayel, Kieu-Dung Ly, K. V. Raghavendra Rao, Gael De Paëpe, Katharina Märker, Anne Milet (2017), "At the very beginning of life on Earth: the thiol-rich peptide (TRP) world hypothesis", The International Journal of Developmental Biology 61 (8–9): 471–478, doi:10.1387/ijdb.170028yv, PMID 29139533
- ↑ 206.0 206.1 James P. Ferris, William J. Hagan (January 1984), "HCN and chemical evolution: The possible role of cyano compounds in prebiotic synthesis", Tetrahedron 40 (7): 1093–1120, doi:10.1016/S0040-4020(01)99315-9, PMID 11541961, Bibcode: 1984Tetra..40.1093F
- ↑ L. Chimiak, J. Eiler, A. Sessions, C. Blumenfeld, M. Klatte, B.M. Stoltz (March 2022), "Isotope effects at the origin of life: Fingerprints of the Strecker synthesis", Geochimica et Cosmochimica Acta 321: 78–98, doi:10.1016/j.gca.2022.01.015, Bibcode: 2022GeCoA.321...78C
- ↑ Ibrahim Shalayel, Seydou Coulibaly, Kieu Ly, Anne Milet, Yannick Vallée (2018-10-19), "The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean", Life 8 (4): 47, doi:10.3390/life8040047, PMID 30347745, Bibcode: 2018Life....8...47S
- ↑ 209.0 209.1 209.2 "Hydrogen Cyanide", Synthetic Nitrogen Products (Boston: Kluwer Academic Publishers) 19: 347–360, 2005, doi:10.1007/0-306-48639-3_19, ISBN 0-306-48225-8
- ↑ "Ethylenediamine and Chelating Agents", Synthetic Nitrogen Products (Boston: Kluwer Academic Publishers) 16: 325–331, 2005, doi:10.1007/0-306-48639-3_16, ISBN 0-306-48225-8
- ↑ Harald Gröger (2003-08-01), "Catalytic Enantioselective Strecker Reactions and Analogous Syntheses", Chemical Reviews 103 (8): 2795–2828, doi:10.1021/cr020038p, PMID 12914481
- ↑ Thomas Willke (December 2014), "Methionine production—a critical review", Applied Microbiology and Biotechnology 98 (24): 9893–9914, doi:10.1007/s00253-014-6156-y, PMID 25381187
- ↑ 213.0 213.1 Arman Sedghi, Reza Eslami Farsani, Ali Shokuhfar (March 2008), "The effect of commercial polyacrylonitrile fibers characterizations on the produced carbon fibers properties", Journal of Materials Processing Technology 198 (1–3): 60–67, doi:10.1016/j.jmatprotec.2007.06.052
- ↑ Md Abdullah Al Faruque, Rechana Remadevi, Joselito Razal, Xungai Wang, Maryam Naebe (2020-02-15), "Investigation on structure and characteristics of alpaca-based wet-spun polyacrylonitrile composite fibers by utilizing natural textile waste", Journal of Applied Polymer Science 137 (7), doi:10.1002/app.48370, https://figshare.com/articles/journal_contribution/20745766
- ↑ B. E. Geller (2002), "Status and Prospects for Development of Polyacrylonitrile Fibre Production. A Review", Fibre Chemistry 34 (3): 151–161, doi:10.1023/A:1020525628197
- ↑ P. Bajaj, Surya Kumari (May 1987), "Modification of Acrylic Fibers: An Overview", Journal of Macromolecular Science, Part C: Polymer Reviews 27 (2): 181–217, doi:10.1080/07366578708081915
- ↑ T.A. Adegbola, O. Agboola, O.S.I. Fayomi (September 2020), "Review of polyacrylonitrile blends and application in manufacturing technology: recycling and environmental impact", Results in Engineering 7, doi:10.1016/j.rineng.2020.100144
- ↑ Xing Jin, Chunfang Feng, Claudia Creighton, Nishar Hameed, Jyotishkumar Parameswaranpillai, Nisa V. Salim (2021-04-01), "On the structural evolution of textile grade polyacrylonitrile fibers during stabilization and carbonization: Towards the manufacture of low-cost carbon fiber", Polymer Degradation and Stability 186, doi:10.1016/j.polymdegradstab.2021.109536, https://figshare.com/articles/journal_contribution/26252870
- ↑ J. Sawyer (2005-12-12), "Comparing the Level of Dexterity offered by Latex and Nitrile SafeSkin Gloves", Annals of Occupational Hygiene 50 (3): 289–296, doi:10.1093/annhyg/mei066, PMID 16357028
- ↑ Keh-Ping Chao, Pak-Hing Lee, Min-Jet Wu (April 2003), "Organic solvents permeation through protective nitrile gloves", Journal of Hazardous Materials 99 (2): 191–201, doi:10.1016/S0304-3894(03)00042-6, PMID 12719151, Bibcode: 2003JHzM...99..191C
- ↑ L. B. Brennan, D. H. Isaac, J. C. Arnold (2002-10-17), "Recycling of acrylonitrile–butadiene–styrene and high-impact polystyrene from waste computer equipment", Journal of Applied Polymer Science 86 (3): 572–578, doi:10.1002/app.10833, Bibcode: 2002JAPS...86..572B
- ↑ 222.0 222.1 "Acrylonitrile-butadiene-styrene copolymers", Encyclopedic Dictionary of Polymers (New York, NY: Springer New York): 22, 2006
- ↑ Francesca Sabatini, Silvia Pizzimenti, Irene Bargagli, Ilaria Degano, Celia Duce, Laura Cartechini, Francesca Modugno, Francesca Rosi (2023-07-31), "A Thermal Analytical Study of LEGO® Bricks for Investigating Light-Stability of ABS", Polymers 15 (15): 3267, doi:10.3390/polym15153267, PMID 37571161
- ↑ Zhijuan Wang, Hongyan Li, Tao Li, Qing Zhang, Yaqi Cai, Hua Bai, Qing Lv (March 2023), "Application of validated migration models for the risk assessment of styrene and acrylonitrile in ABS plastic toys", Ecotoxicology and Environmental Safety 252, doi:10.1016/j.ecoenv.2023.114570, PMID 36706528, Bibcode: 2023EcoES.25214570W
- ↑ Alex Tullo (2018-10-08), "Industry braces for nylon 6,6 shortage", C&EN Global Enterprise 96 (40): 22–23, doi:10.1021/cen-09640-feature3
- ↑ Younghyun Lee, Sung Woo Lee, Hyung Ju Kim, Yong Tae Kim, Kun-Yi Andrew Lin, Jechan Lee (2020-10-26), "Hydrogenation of Adiponitrile to Hexamethylenediamine over Raney Ni and Co Catalysts", Applied Sciences 10 (21): 7506, doi:10.3390/app10217506
- ↑ Mohammad Jaber Darabi Mahboub, Jean-Luc Dubois, Fabrizio Cavani, Mohammad Rostamizadeh, Gregory S. Patience (2018), "Catalysis for the synthesis of methacrylic acid and methyl methacrylate", Chemical Society Reviews 47 (20): 7703–7738, doi:10.1039/C8CS00117K, PMID 30211916
- ↑ 228.0 228.1 Ian F. McConvey, Dean Woods, Moira Lewis, Quan Gan, Paul Nancarrow (2012-04-20), "The Importance of Acetonitrile in the Pharmaceutical Industry and Opportunities for its Recovery from Waste", Organic Process Research & Development 16 (4): 612–624, doi:10.1021/op2003503, https://pure.qub.ac.uk/en/publications/de533647-f193-49f2-b6e7-9eee75cf765f
- ↑ "Acetonitrile Market Size, Growth, Analysis & Forecast, 2032". https://www.chemanalyst.com/industry-report/acetonitrile-market-721.
- ↑ Cristiano Soleo Funari, Renato Lajarim Carneiro, Manish M. Khandagale, Alberto José Cavalheiro, Emily F. Hilder (May 2015), "Acetone as a greener alternative to acetonitrile in liquid chromatographic fingerprinting", Journal of Separation Science 38 (9): 1458–1465, doi:10.1002/jssc.201401324, PMID 25708832
- ↑ Ladislav Androvič, Jan Bartáček, Miloš Sedlák (June 2016), "Recent advances in the synthesis and applications of azo initiators", Research on Chemical Intermediates 42 (6): 5133–5145, doi:10.1007/s11164-015-2351-4
- ↑ Bao Li, Alison E. Wendlandt, Shannon S. Stahl (2019-02-15), "Replacement of Stoichiometric DDQ with a Low Potential o -Quinone Catalyst Enabling Aerobic Dehydrogenation of Tertiary Indolines in Pharmaceutical Intermediates", Organic Letters 21 (4): 1176–1181, doi:10.1021/acs.orglett.9b00111, PMID 30702297
- ↑ Jun-Ho Choi, Kwang-Im Oh, Hochan Lee, Chewook Lee, Minhaeng Cho (2008-04-07), "Nitrile and thiocyanate IR probes: Quantum chemistry calculation studies and multivariate least-square fitting analysis", The Journal of Chemical Physics 128 (13), doi:10.1063/1.2844787, PMID 18397076, Bibcode: 2008JChPh.128m4506C
- ↑ Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz (1991), Ullmann's encyclopedia of industrial chemistry (5th ed.), Weinheim New York: VCH, ISBN 3-527-20117-3
- ↑ S. DAWSON (2005-08-01), THE HIGGS WORKING GROUP: SUMMARY REPORT., Office of Scientific and Technical Information (OSTI)
- ↑ Lili Song, Zhigang Liu, Minjie Liu, Pei Tang, Fener Chen (2023-05-19), "Efficient and Scalable Synthesis of Ketoprofen: A Pyrolytic Aromatization Approach", Organic Process Research & Development 27 (5): 922–927, doi:10.1021/acs.oprd.3c00049
- ↑ 237.0 237.1 Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1650–1671, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ 238.0 238.1 238.2 Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1651, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1650–1671, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1651f, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ Ricky C.K. Cheng, Denis B. Tikhonov, Boris S. Zhorov (October 2009), "Structural Model for Phenylalkylamine Binding to L-type Calcium Channels", Journal of Biological Chemistry 284 (41): 28332–28342, doi:10.1074/jbc.M109.027326, PMID 19700404
- ↑ Natalija Popović, Nicanor Morales-Delgado, David Vidal Mena, Antonia Alonso, María Pascual Martínez, María Caballero Bleda, Miroljub Popović (2020-05-05), "Verapamil and Alzheimer's Disease: Past, Present, and Future", Frontiers in Pharmacology 11, doi:10.3389/fphar.2020.00562, PMID 32431612
- ↑ Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1654, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ Junmei Cairns, James N. Ingle, Tanda M. Dudenkov (2020-08-20), "Pharmacogenomics of aromatase inhibitors in postmenopausal breast cancer and additional mechanisms of anastrozole action", JCI Insight 5 (16), doi:10.1172/jci.insight.137571, PMID 32701512
- ↑ Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT (2005). "Structural basis for antagonism and resistance of bicalutamide in prostate cancer". Proc Natl Acad Sci U S A 102 (17): 6201–6. doi:10.1073/pnas.0500381102. PMID 15833816. Bibcode: 2005PNAS..102.6201B.
- ↑ Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1653, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ Mahnoor Pasha, Ammara Zamir, Waseem Ashraf, Imran Imran, Hamid Saeed, Anees Ur Rehman, Majid Aziz, Faleh Alqahtani, Muhammad Fawad Rasool (2023-11-26), "A systematic review on the clinical pharmacokinetics of vildagliptin in healthy and disease populations", Expert Opinion on Drug Metabolism & Toxicology 19 (12): 991–1003, doi:10.1080/17425255.2023.2288252, PMID 38008954
- ↑ Mika Nabeno, Fumihiko Akahoshi, Hiroyuki Kishida, Ikuko Miyaguchi, Yoshihito Tanaka, Shinichi Ishii, Takashi Kadowaki (May 2013), "A comparative study of the binding modes of recently launched dipeptidyl peptidase IV inhibitors in the active site", Biochemical and Biophysical Research Communications 434 (2): 191–196, doi:10.1016/j.bbrc.2013.03.010, PMID 23501107, Bibcode: 2013BBRC..434..191N
- ↑ Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du (2021), "Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies", RSC Medicinal Chemistry 12 (10): 1660, doi:10.1039/D1MD00131K, PMID 34778767
- ↑ Sandra E. Pineda-Sanabria, Ian M. Robertson, Yin-Biao Sun, Malcolm Irving, Brian D. Sykes (March 2016), "Probing the mechanism of cardiovascular drugs using a covalent levosimendan analog", Journal of Molecular and Cellular Cardiology 92: 174–184, doi:10.1016/j.yjmcc.2016.02.003, PMID 26853943
- ↑ Das K, Bauman JD, Clark AD, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E (2008). "High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations". Proc Natl Acad Sci U S A 105 (5): 1466–71. doi:10.1073/pnas.0711209105. PMID 18230722.
- ↑ Coleman JA, Green EM, Gouaux E (2016). "X-ray structures and mechanism of the human serotonin transporter". Nature 532 (7599): 334–9. doi:10.1038/nature17629. PMID 27049939. Bibcode: 2016Natur.532..334C.
- ↑ Fleming, Fraser F.; Yao, Lihua; Ravikumar, P. C.; Funk, Lee; Shook, Brian C. (November 2010). "Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore". J Med Chem 53 (22): 7902–17. doi:10.1021/jm100762r. PMID 20804202. Bibcode: 2010JMedC..53.7902F.
- ↑ David R Bickers, Peter Calow, Helmut A Greim, Jon M Hanifin, Adrianne E Rogers, Jean-Hilaire Saurat, I Glenn Sipes, Robert L Smith, Hachiro Tagami (April 2003), "The safety assessment of fragrance materials", Regulatory Toxicology and Pharmacology 37 (2): 218–273, doi:10.1016/S0273-2300(03)00003-5, PMID 12726755
- ↑ S.P. Bhatia, V.T. Politano, A.M. Api (September 2013), "Evaluation of genotoxicity of nitrile fragrance ingredients using in vitro and in vivo assays", Food and Chemical Toxicology 59: 784–792, doi:10.1016/j.fct.2013.04.040, PMID 23643699
- ↑ K.R. Brain, D.M. Green, J. Lalko, A.M. Api (February 2007), "In-vitro human skin penetration of the fragrance material geranyl nitrile", Toxicology in Vitro 21 (1): 133–138, doi:10.1016/j.tiv.2006.08.005, PMID 17045775, Bibcode: 2007ToxVi..21..133B
- ↑ David Pybus, Charles Sell (1999), The chemistry of fragrances, RSC paperbacks, Cambridge: Royal Society of Chemistry, p. 66, ISBN 0-85404-528-7
- ↑ J E Casida, D W Gammon, A H Glickman, L J Lawrence (April 1983), "Mechanisms of Selective Action of Pyrethroid Insecticides", Annual Review of Pharmacology and Toxicology 23 (1): 413–438, doi:10.1146/annurev.pa.23.040183.002213, PMID 6347050
- ↑ Thomas C. Sparks, James E. Hunter, Beth A. Lorsbach, Greg Hanger, Roger E. Gast, Greg Kemmitt, Robert J. Bryant (2018-10-10), "Crop Protection Discovery: Is Being the First Best?", Journal of Agricultural and Food Chemistry 66 (40): 10337–10346, doi:10.1021/acs.jafc.8b03484, PMID 30205003, Bibcode: 2018JAFC...6610337S
- ↑ 260.0 260.1 Dave W Bartlett, John M Clough, Jeremy R Godwin, Alison A Hall, Mick Hamer, Bob Parr-Dobrzanski (July 2002), "The strobilurin fungicides", Pest Management Science 58 (7): 649–662, doi:10.1002/ps.520, PMID 12146165, Bibcode: 2002PMSci..58..649B
- ↑ Ngangbam Sarat Singh, Ranju Sharma, Sandeep Kumar Singh, Dileep Kumar Singh (August 2021), "A comprehensive review of environmental fate and degradation of fipronil and its toxic metabolites", Environmental Research 199, doi:10.1016/j.envres.2021.111316, PMID 33989624, Bibcode: 2021ER....19911316S
- ↑ H. W. Coover, D. W. Dreifus, J. T. O'Connor (1990), "Cyanoacrylate Adhesives", Handbook of Adhesives (Boston, MA: Springer US): 463–477, doi:10.1007/978-1-4613-0671-9_27, ISBN 1-4612-8019-2
- ↑ "Wie ein gewöhnlicher Zusatzstoff Lithium-Ionen-Batterien einen Schub verleiht". Chemie.de. 2022-09-28. https://www.chemie.de/news/1177889/wie-ein-gewoehnlicher-zusatzstoff-lithium-ionen-batterien-einen-schub-verleiht.html?xing_share=news.
- ↑ Chao Tang, Yawei Chen, Zhengfeng Zhang, Wenqiang Li, Junhua Jian, Yulin Jie, Fanyang Huang, Yehu Han, Wanxia Li, Fuping Ai, Ruiguo Cao, Pengfei Yan, Yuhao Lu, Shuhong Jiao (2022), "Stable cycling of practical high-voltage LiCoO2 pouch cell via electrolyte modification", Nano Research 16 (3): 3864–3871, doi:10.1007/s12274-022-4955-5
External links
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "nitrile". doi:10.1351/goldbook.N04151
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "cyanide". doi:10.1351/goldbook.C01486
 |  |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
_fruits_(29055398276).jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
-mandelonitrile-2D-skeletal.svg){kind=link}
_The_Magpie_(Abraxas_grossulariata)_(14502341918).jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}