Chemistry:Homoaromaticity
Homoaromaticity, in organic chemistry, refers to a special case of aromaticity in which conjugation is interrupted by a single sp3 hybridized carbon atom. Although this sp3 center disrupts the continuous overlap of p-orbitals, traditionally thought to be a requirement for aromaticity, considerable thermodynamic stability and many of the spectroscopic, magnetic, and chemical properties associated with aromatic compounds are still observed for such compounds. This formal discontinuity is apparently bridged by p-orbital overlap, maintaining a contiguous cycle of π electrons that is responsible for this preserved chemical stability.[1]

The concept of homoaromaticity was pioneered by Saul Winstein in 1959, prompted by his studies of the “tris-homocyclopropenyl” cation.[2] Since the publication of Winstein's paper, much research has been devoted to understanding and classifying these molecules, which represent an additional class of aromatic molecules included under the continuously broadening definition of aromaticity. To date, homoaromatic compounds are known to exist as cationic and anionic species, and some studies support the existence of neutral homoaromatic molecules, though these are less common.[3] The 'homotropylium' cation (C8H9+) is perhaps the best studied example of a homoaromatic compound.
Overview
Naming
The term "homoaromaticity" derives from the structural similarity between homoaromatic compounds and the analogous homo-conjugated alkenes previously observed in the literature.[2] The IUPAC Gold Book requires that Bis-, Tris-, etc. prefixes be used to describe homoaromatic compounds in which two, three, etc. sp3 centers separately interrupt conjugation of the aromatic system.
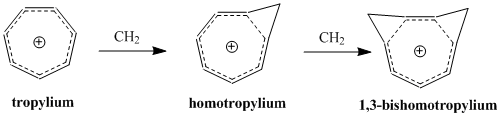
IUPAC naming system illustrated by the homotropylium cation derivatives

History
The concept of homoaromaticity has its origins in the debate over the non-classical carbonium ions that occurred in the 1950s. Saul Winstein, a famous proponent of the non-classical ion model, first described homoaromaticity while studying the 3-bicyclo[3.1.0]hexyl cation.

Trishomoaromaticity

In a series of acetolysis experiments, Winstein et al. observed that the solvolysis reaction occurred empirically faster when the tosyl leaving group was in the equatorial position. The group ascribed this difference in reaction rates to the anchimeric assistance invoked by the "cis" isomer. This result thus supported a non-classical structure for the cation.[4]
Winstein subsequently observed that this non-classical model of the 3-bicyclo[3.1.0]hexyl cation is analogous to the previously well-studied aromatic cyclopropenyl cation. Like the cyclopropenyl cation, positive charge is delocalized over three equivalent carbons containing two π electrons. This electronic configuration thus satisfies Huckel's rule (requiring 4n+2 π electrons) for aromaticity. Indeed, Winstein noticed that the only fundamental difference between this aromatic propenyl cation and his non-classical hexyl cation was the fact that, in the latter ion, conjugation is interrupted by three -CH2- units. The group thus proposed the name "tris-homocyclopropenyl"—the tris-homo counterpart to the cyclopropenyl cation.
Evidence for homoaromaticity
Criterion for homoaromaticity
The criterion for aromaticity has evolved as new developments and insights continue to contribute to our understanding of these remarkably stable organic molecules.[5] The required characteristics of these molecules has thus remained the subject of some controversy. Classically, aromatic compounds were defined as planar molecules that possess a cyclically delocalized system of (4n+2)π electrons, satisfying Huckel's rule. Most importantly, these conjugated ring systems are known to exhibit enormous thermochemical stability relative to predictions based on localized resonance structures. Succinctly, three important features seem to characterize aromatic compounds:[6]
- molecular structure (i.e. coplanarity: all contributing atoms in the same plane)
- molecular energetics (i.e. increased thermodynamic stability)
- spectroscopic and magnetic properties (i.e. magnetic field induced ring current)
A number of exceptions to these conventional rules exist, however. Many molecules, including Möbius 4nπ electron species, pericyclic transition states, molecules in which delocalized electrons circulate in the ring plane or through σ (rather than π) bonds, many transition-metal sandwich molecules, and others have been deemed aromatic though they somehow deviate from the conventional parameters for aromaticity.[7]
Consequently, the criterion for homoaromatic delocalization remains similarly ambiguous and somewhat controversial. The homotropylium cation, (C8H9+), though not the first example of a homoaromatic compound ever discovered, has proven to be the most studied of the compounds classified as homoaromatic, and is therefore often considered the classic example of homoaromaticity. By the mid-1980s, there were more than 40 reported substituted derivatives of the homotropylium cation, reflecting the importance of this ion in formulating our understanding of homoaromatic compounds.[6]
Early evidence for homoaromaticity
After initial reports of a "homoaromatic" structure for the tris-homocyclopropenyl cation were published by Winstein, many groups began to report observations of similar compounds. One of the best studied of these molecules is the homotropylium cation, the parent compound of which was first isolated as a stable salt by Pettit, et al. in 1962, when the group reacted cyclooctatraene with strong acids.[8] Much of the early evidence for homoaromaticity comes from observations of unusual NMR properties associated with this molecule.
NMR spectroscopy studies
While characterizing the compound resulting from deprotonation of cyclooctatriene by 1H NMR spectroscopy, the group observed that the resonance corresponding to two protons bonded to the same methylene bridge carbon exhibited an astonishing degree of separation in chemical shift.
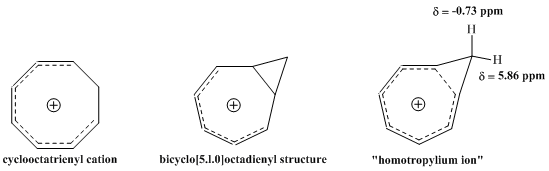
Progression in understanding of the structure of the homotropylium ion.

From this observation, Pettit, et al. concluded that the classical structure of the cyclooctatrienyl cation must be incorrect. Instead, the group proposed the structure of the bicyclo[5.1.0]octadienyl compound, theorizing that the cyclopropane bond located on the interior of the eight-membered ring must be subject to considerable delocalization, thus explaining the dramatic difference in observed chemical shift. Upon further consideration, Pettit was inclined to represent the compound as the "homotropylium ion," which shows the "internal cyclopropane" bond totally replaced by electron delocalization. This structure shows how delocalization is cyclic and involves 6 π electrons, consistent with Huckel's rule for aromaticity. The magnetic field of the NMR could thus induce a ring current in the ion, responsible for the significant differences in resonance between the exo and endo protons of this methylene bridge. Pettit, et al. thus emphasized the remarkable similarity between this compound and the aromatic tropylium ion, describing a new "homo-counterpart" to an aromatic species already known, precisely as predicted by Winstein.

Subsequent NMR studies undertaken by Winstein and others sought to evaluate the properties of metal carbonyl complexes with the homotropylium ion. Comparison between a molybdenum-complex and an iron-complex proved particularly fruitful. Molybdenum tricarbonyl was expected to coordinate to the homotropylium cation by accepting 6 π electrons, thereby preserving the homoaromatic features of the complex. By contrast, iron tricarbonyl was expected to coordinate to the cation by accepting only 4 π electrons from the homotropylium ion, creating a complex in which the electrons of the cation are localized. Studies of these complexes by 1H NMR spectroscopy showed a large difference in chemical shift values for methylene protons of the Mo-complex, consistent with a homoaromatic structure, but detected virtually no comparable difference in resonance for the same protons in the Fe-complex.[9]
UV spectroscopy studies
An important piece of early evidence in support of the homotropylium cation structure that did not rely on the magnetic properties of the molecule involved the acquisition of its UV spectrum. Winstein et al. determined that the absorption maxima for the homotropylium cation exhibited a considerably shorter wavelength than would be precited for the classical cyclooctatrienyl cation or the bicyclo[5.1.0]octadienyl compound with the fully formed internal cyclopropane bond (and a localized electronic structure). Instead, the UV spectrum most resembled that of the aromatic tropylium ion. Further calculations allowed Winstein to determine that the bond order between the two carbon atoms adjacent to the outlying methylene bridge is comparable to that of the π-bond separating the corresponding carbon atoms in the tropylium cation.[10] Although this experiment proved to be highly illuminating, UV spectra are generally considered to be poor indicators of aromaticity or homoaromaticity.[6]
More recent evidence for homoaromaticity
More recently, work has been done to investigate the structure of the purportedly homoaromatic homotropylium ion by employing various other experimental techniques and theoretical calculations. One key experimental study involved analysis of a substituted homotropylium ion by X-ray crystallography. These crystallographic studies have been used to demonstrate that the internuclear distance between the atoms at the base of the cyclopropenyl structure is indeed longer than would be expected for a normal cyclopropane molecule, while the external bonds appear to be shorter, indicating involvement of the internal cyclopropane bond in charge delocalization.[6]
Molecular orbital description
The molecular orbital explanation of the stability of homoaromaticity has been widely discussed with numerous diverse theories, mostly focused on the homotropenylium cation as a reference. R.C. Haddon initially proposed a Mobius model where the outer electrons of the sp3 hybridized methylene bridge carbon(2) back-donate to the adjacent carbons to stabilize the C1-C3 distance.[11]
Perturbation molecular orbital theory
Homoaromaticity can better be explained using Perturbation Molecular Orbital Theory (PMO) as described in a 1975 study by Robert C. Haddon. The homotropenylium cation can be considered as a perturbed version of the tropenylium cation due to the addition of a homoconjugate linkage interfering with the resonance of the original cation.[12]

First-order effects
The most important factor in influencing homoaromatic character is the addition of a single homoconjugate linkage into the parent aromatic compound. The location of the homoconjugate bond is not important as all homoaromatic species can be derived from aromatic compounds that possess symmetry and equal bond order between all carbons. The insertion of a homoconjugate linkage perturbs the π-electron density an amount δβ, which depending on the ring size, must be greater than 0 and less than 1, where 0 represents no perturbation and 1 represents total loss of aromaticity (destabilization equivalent to the open chain form).[12] It is believed that with increasing ring size, the resonance stabilization of homoaromaticity is offset by the strain in forming the homoconjugate bridge. In fact, the maximum ring size for homoaromaticity is fairly low as a 16-membered annulene ring favours the formation of the aromatic dication over the strained bridged homocation.[13]
Second-order effects
Second homoconjugate linkage
A significant second-order effect on the Perturbation Molecular Orbital model of homoaromaticity is the addition of a second homoconjugate linkage and its influence on stability. The effect is often a doubling of the instability brought about by the addition of a single homoconjugate linkage, although there is an additional term that depends on the proximity of the two linkages. In order to minimize δβ and thus keep the coupling term to a minimum, bishomoaromatic compounds form depending on the conformation of greatest stability by resonance and smallest steric hindrance. The synthesis of the 1,3-bishomotropenylium cation by protonating cis-bicyclo[6.1.0]nona-2,4,6-triene agrees with theoretical calculations and maximizes stability by forming the two methylene bridges at the 1st and 3rd carbons.[12]

Substituents
The addition of a substituent to a homoaromatic compound has a large influence over the stability of the compound. Depending on the relative locations of the substituent and the homoconjugate linkage, the substituent can either have a stabilizing or destabilizing effect. This interaction is best demonstrated by looking at a substituted tropenylium cation. If an inductively electron-donating group is attached to the cation at the 1st or 3rd carbon position, it has a stabilizing effect, improving the homoaromatic character of the compound. However, if this same substituent is attached at the 2nd or 4th carbon, the interaction between the substituent at the homoconjugate bridge has a destabilizing effect. Therefore, protonation of methyl or phenyl substituted cyclooctatetraenes will result in the 1 isomer of the homotropenylium cation.[12]

Examples of homoaromatic compounds
Following the discovery of the first homoaromatic compounds, research has gone into synthesizing new homoaromatic compounds that possess similar stability to their aromatic parent compounds. There are several classes of homoaromatic compounds, each of which have been predicted theoretically and proven experimentally.
Cationic homoaromatics
The most established and well-known homoaromatic species are cationic homoaromatic compounds. As stated earlier, the homotropenylium cation is one of the most studied homoaromatic compounds. Many homoaromatic cationic compounds use as a basis a cyclopropenyl cation, a tropylium cation, or a cyclobutadiene dication as these compounds exhibit strong aromatic character.[14]

In addition to the homotropylium cation, another well established cationic homoaromatic compound is the norbornen-7-yl cation, which has been shown to be strongly homoaromatic, proven both theoretically and experimentally.[15]

An intriguing case of σ-bishomoaromaticity can be found in the dications of pagodanes. In these 4-center-2-electron systems the delocalization happens in the plane that is defined by the four carbon atoms (prototype for the phenomenon of σ-aromaticity is cyclopropane which gains about 11.3 kcal mol−1 stability from the effect[16]). The dications are accessible either via oxidation of pagodane or via oxidation of the corresponding bis-seco-dodecahedradiene:[17]
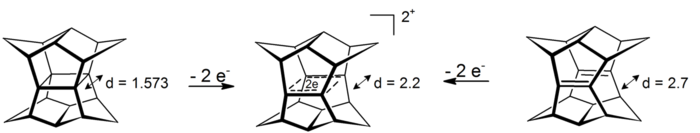
Oxidation of pagodane and dodecahedradiene to a sigma-bishomoaromatic dication. Distances in angstrom calculated at HF/3-21G level for the dication an the diene, x-ray for the neutral

Reduction of the corresponding six electrons dianions was not possible so far.
Neutral homoaromatics
There are many classes of neutral homoaromatic compounds although there is much debate as to whether they truly exhibit homoaromatic character or not.
One class of neutral homoaromatics are called monohomoaromatics, one of which is cycloheptatriene, and numerous complex monohomoaromatics have been synthesized. One particular example is a 60-carbon fulleroid derivative that has a single methylene bridge. UV and NMR analysis have shown that the aromatic character of this modified fulleroid is not disrupted by the addition of a homoconjugate linkage, therefore this compound is definitively homoaromatic.[18]
Substituted neutral barbaralane derivatives (homoannulenes) have been disclosed as stable ground state homoaromatic molecules in 2023. Evidence for the homoaromatic character in this class of molecules stems from bond length analysis (X-Ray structural analysis) as well as shifts in the NMR spectrum.[19][20] The homoannulenes also act as photoswitches by which means a local 6π homoaromaticity can be switched to a global 10π homoaromaticity.

Bishomoaromatics
It was long considered that the best examples of neutral homoaromatics are bishomoaromatics such as barrelene and semibullvalene. First synthesized in 1966,[21] semibullvalene has a structure that should lend itself well to homoaromaticity although there has been much debate whether semibullvalene derivatives can provide a true delocalized, ground state neutral homoaromatic compound or not. In an effort to further stabilize the delocalized transition structure by substituting semibullvalene with electron donating and accepting groups, it has been found that the activation barrier to this rearrangement can be lowered, but not eliminated.[22][23] However, with the introduction of ring strain into the molecule, aimed at destabilizing the localized ground state structure's through the strategic addition of cyclic annulations, a delocalized homoaromatic ground-state structure can indeed be achieved.[24]

Of the neutral homoaromatics, the compounds best believed to exhibit neutral homoaromaticity are boron containing compounds of 1,2-diboretane and its derivatives. Substituted diboretanes are shown to have a much greater stabilization in the delocalized state over the localized one, giving strong indications of homoaromaticity.[25] When electron-donating groups are attached to the two boron atoms, the compound favors a classical model with localized bonds. Homoaromatic character is best seen when electron-withdrawing groups are bonded to the boron atoms, causing the compound to adopt a nonclassical, delocalized structure.

Trishomoaromatics
As the name suggests, trishomoaromatics are defined as containing one additional methylene bridge compared to bishomoaromatics, therefore containing three of these homoconjugate bridges in total. Just like semibullvalene, there is still much debate as to the extent of the homoaromatic character of trishomoaromatics. While theoretically they are homoaromatic, these compounds show a stabilization of no more than 5% of benzene due to delocalization.[26]

Anionic homoaromatics
Unlike neutral homoaromatic compounds, anionic homoaromatics are widely accepted to exhibit "true" homoaromaticity. These anionic compounds are often prepared from their neutral parent compounds through lithium metal reduction. 1,2-diboretanide derivatives show strong homoaromatic character through their three-atom (boron, boron, carbon), two-electron bond, which contains shorter C-B bonds than in the neutral classical analogue.[27] These 1,2-diboretanides can be expanded to larger ring sizes with different substituents and all contain some degree of homoaromaticity.

Anionic homoaromaticity can also be seen in dianionic bis-diazene compounds, which contain a four-atom (four nitrogens), six-electron center. Experiment results have shown the shortening of the transannular nitrogen-nitrogen distance, therefore demonstrating that dianionic bis-diazene is a type of anionic bishomoaromatic compound. Peculiar feature of these systems is that the cyclic electron delocalization is taking place in the σ-plane defined by the four nitrogens. These bis-diazene-dianions are therefore the first examples for 4-center-6-electron σ-bishomoaromaticity.[28][29] The corresponding 2 electron σ-bishomoaromatic systems were realized in the form of pagodane dications (see above).
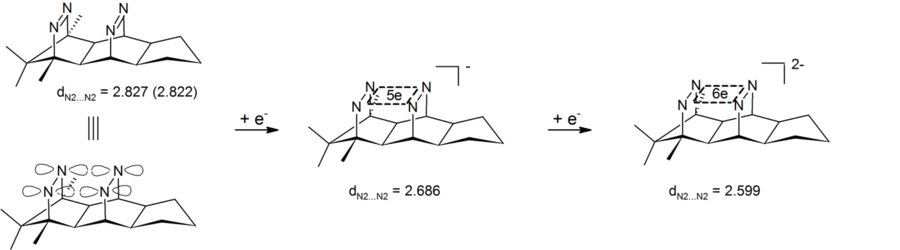
Reduction of a bisdiazene to a sigma-bishomoaromatic dianion. Distances in angstrom calculated at B3LYP?6-31G* level (x-ray for the neutral)

Antihomoaromaticity
There are also reports of antihomoaromatic compounds. Just as aromatic compounds exhibit exceptional stability, antiaromatic compounds, which deviate from Huckel's rule and contain a closed loop of 4n π electrons, are relatively unstable. The bridged bicyclo[3.2.1]octa-3,6-dien-2-yl cation contains only 4 π electrons, and is therefore "bishomoantiaromatic." A series of theoretical calculations confirm that it is indeed less stable than the corresponding allyl cation.[30]

Similarly, a substituted bicyclo[3.2.1]octa-3,6-dien-2-yl cation (the 2-(4'-Fluorophenyl) bicyclo[3.2.1]oct-3,6-dien-2-yl cation) was also shown to be an antiaromate when compared to its corresponding allyl cation, corroborated by theoretical calculations as well as by NMR analysis.[30]

External links
- Homoaromaticity on the Gold Book
References
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Homoaromatic". doi:10.1351/goldbook.H02839
- ↑ 2.0 2.1 Winstein, S (1959). "Homo-Aromatic Structures". J. Am. Chem. Soc. 81 (24): 6523. doi:10.1021/ja01533a052.
- ↑ Freeman, P. K. (2005). "Neutral Homoaromaticity in Some Neutral Heterocycles". J. Org. Chem. 70 (6): 1998–2001. doi:10.1021/jo040250o. PMID 15760178.
- ↑ Winstein, S.; Sonnenberg, J.; DeVries, L. (1959). "The Tris-Homocyclopropenyl Cation". J. Am. Chem. Soc. 81 (24): 6523–6524. doi:10.1021/ja01533a051.
- ↑ le Noble, W. J. "Aromaticity" in Highlights of Organic Chemistry: an advanced textbook; Marcel Dekker, Inc.: New York, 1974. ISBN 0-8247-6210-X
- ↑ 6.0 6.1 6.2 6.3 Childs, R. F. (1984). "The Homotropylium Ion and Homoaromaticity". Acc. Chem. Res. 17 (10): 347–352. doi:10.1021/ar00106a001.
- ↑ Schleyer, P. R. (2001). "Introduction: Aromaticity". Chem. Rev. 101 (5): 1115–1118. doi:10.1021/cr0103221. PMID 11749368.
- ↑ Rosenburg, J. L.; Mahler, J. E.; Pettit, R. J. (1962). "The Bicyclo[5.1.0]octadienyl Cation, A New Stable Carbonium Ion". J. Am. Chem. Soc. 84 (14): 2842–2843. doi:10.1021/ja00873a051.
- ↑ Winstein, S.; Kaesz, H.D.; Kreiter, C.G.; Friedrich, E.C. (1965). "Homotropylium Ion and its Molybdenum Tricarbonyl Complex". J. Am. Chem. Soc. 87 (14): 3267–3269. doi:10.1021/ja01092a060.
- ↑ Winstein, S.; Kreiter, C.G.; Brauman, J.I. (1966). "Ring Inversion, Ultraviolet Spectrum, and Electronic Structure of the Monohomotropylium Ion". J. Am. Chem. Soc. 88 (9): 2047–2048. doi:10.1021/ja00961a037.
- ↑ Haddon, R.C. (1975). "The structure of the homotropenylium cation". Tetrahedron Lett. 16 (11): 863–866. doi:10.1016/S0040-4039(00)72004-1.
- ↑ 12.0 12.1 12.2 12.3 Haddon, R.C. (1975). "Perturbational molecular orbital (PMO) theory of homoaromaticity". J. Am. Chem. Soc. 97 (13): 3608–3615. doi:10.1021/ja00846a009.
- ↑ Oth, J.F.M.; Smith, D.M.; Prange, U.; Schröder, G. (1973). "A [16]Annulenediyl Dication". Angew. Chem. Int. Ed. Engl. 12 (4): 327–328. doi:10.1002/anie.197303271.
- ↑ Sal'nikov, G.E.; Genaev, A.M.; Mamatyuk, V.I.; Shubin, V.G. (2008). "Homophenalenyl cations, new representatives of homoaromatic systems". Russ. J. Org. Chem. 44 (7): 1000–1005. doi:10.1134/S1070428008070099.
- ↑ Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry: Part A: Structure and Mechanism; Kluwer Academic/Plenum Publishers: New York, 2000; 327-334. ISBN 978-0-387-68346-1
- ↑ Exner, Kai; Schleyer, Paul von Ragué (2001). "Theoretical Bond Energies: A Critical Evaluation". J. Phys. Chem. A 105 (13): 3407–3416. doi:10.1021/jp004193o. Bibcode: 2001JPCA..105.3407E.
- ↑ Prinzbach, H.; Gescheidt, G.; Martin, H.-D.; Herges, R.; Heinze, J.; Prakash, G. K. Surya; Olah, G. A.. "Cyclic electron delocalization in hydrocarbon cages (pagodanes, isopagodanes, (bisseco-/seco-)-(dodecahedradienes))". Pure and Applied Chemistry 67 (5): 673–682, 1995. doi:10.1351/pac199567050673.
- ↑ Suzuki, T.; Li, Q.; Khemani, K.C.; Wudl, F. (1992). "Dihydrofulleroid H3C61: synthesis and properties of the parent fulleroid". J. Am. Chem. Soc. 114 (18): 7301–7302. doi:10.1021/ja00044a055.
- ↑ Tran Ngoc, Trung; Grabicki, Niklas; Irran, Elisabeth; Dumele, Oliver; Teichert, Johannes F. (March 2023). "Photoswitching neutral homoaromatic hydrocarbons" (in en). Nature Chemistry 15 (3): 377–385. doi:10.1038/s41557-022-01121-w. ISSN 1755-4349. PMID 36702883. PMC 9986110. https://www.nature.com/articles/s41557-022-01121-w.
- ↑ Tran Ngoc, Trung; van der Welle, Jasper; Rüffer, Tobias; Teichert, Johannes F. (2023-07-03). "Synthesis of Stable Neutral Homoaromatic Hydrocarbons" (in en). Synthesis. doi:10.1055/s-0042-1751468. ISSN 0039-7881. http://www.thieme-connect.de/DOI/DOI?10.1055/s-0042-1751468.
- ↑ Zimmerman, H. E.; Grunewald, G. L. (1966). "The Chemistry of Barrelene. III. A Unique Photoisomerization to Semibullvalene". Journal of the American Chemical Society 88: 183–184. doi:10.1021/ja00953a045.
- ↑ Dewar, M.J.S.; Lo, D.H. (1971). "Ground states of .sigma.-bonded molecules. XIV. Application of energy partitioning to the MINDO/2 method and a study of the Cope rearrangement". J. Am. Chem. Soc. 93 (26): 7201–7207. doi:10.1021/ja00755a014.
- ↑ Hoffman, D.; Stohrer, W-D (1971). "Cope rearrangement revisited". J. Am. Chem. Soc. 93 (25): 6941–6948. doi:10.1021/ja00754a042.
- ↑ Griffiths, P. R.; Pivonka, D. E.; Williams, R. V. (2011). "The Experimental Realization of a Neutral Homoaromatic Carbocycle". Chemistry: A European Journal 17 (33): 9193–9199. doi:10.1002/chem.201100025. PMID 21735493.
- ↑ Steiner, D.; Balzereit, C.; Winkler, H. J. R.; Stamatis, N.; Massa, W.; Berndt, A.; Hofmann, M.; Von Ragué Schleyer, P. (1994). "Nonclassical 1,2-Diboretanes and 1,2-Diborolanes". Angewandte Chemie International Edition in English 33 (22): 2303–2306. doi:10.1002/anie.199423031.
- ↑ Martin, H.D.; Mayer, B. (1983). "Proximity Effects in Organic Chemistry?The Photoelectron Spectroscopic Investigation of Non-Bonding and Transannular Interactions". Angew. Chem. Int. Ed. Engl. 22 (4): 283–314. doi:10.1002/anie.198302831.
- ↑ Steiner, D.; Winkler, H.; Balzereit, C.; Happel, T.; Hofmann, M.; Subramanian, G.; Schleyer, P.V.R.; Massa, W. et al. (1996). "1,2-Diboretanides: Homoaromatic 2π-Electron Compounds with High Inversion Barriers". Angew. Chem. Int. Ed. Engl. 35 (17): 1990–1992. doi:10.1002/anie.199619901.
- ↑ Exner, K.; Hunkler, D.; Gescheidt, G.; Prinzbach, H. (1998). "Do Nonclassical, Cyclically Delocalized 4N/5e Radical Anions and 4N/6e Dianions Exist? – One- and Two-Electron Reduction of Proximate, Synperiplanar Bis-Diazenes". Angew. Chem. Int. Ed. Engl. 37 (13–14): 1910–1913. doi:10.1002/(SICI)1521-3773(19980803)37:13/14<1910::AID-ANIE1910>3.0.CO;2-D.
- ↑ Exner, K.; Cullmann, O.; Vögtle, M.; Prinzbach, H.; Grossmann, B.; Heinze, J.; Liesum, L.; Bachmann, R. et al. (2000). "Cyclic In-Plane Electron Delocalization (σ-Bishomoaromaticity) in 4N/5e Radical Anions and 4N/6e Dianions – Generation, Structures, Properties, Ion-Pairing, and Calculations". J. Am. Chem. Soc. 122 (43): 10650–10660. doi:10.1021/ja0014943.
- ↑ 30.0 30.1 Volz, H.; Shin, J. (2006). "Bicyclo[3.2.1]octa-3,6-dien-2-yl Cation: A Bishomoantiaromate". J. Org. Chem. 71 (6): 2220–2226. doi:10.1021/jo0515125. PMID 16526766.
 |  |
