Chemistry:Transmetalation
Transmetalation (alt. spelling: transmetallation) is a type of organometallic reaction that involves the transfer of ligands from one metal to another. It has the general form:
- M1–R + M2–R′ → M1–R′ + M2–R
where R and R′ can be, but are not limited to, an alkyl, aryl, alkynyl, allyl, halogen, or pseudohalogen group. The reaction is usually an irreversible process due to thermodynamic and kinetic reasons. Thermodynamics will favor the reaction based on the electronegativities of the metals and kinetics will favor the reaction if there are empty orbitals on both metals.[1] There are different types of transmetalation including redox-transmetalation and redox-transmetalation/ligand exchange. During transmetalation the metal-carbon bond is activated, leading to the formation of new metal-carbon bonds.[2] Transmetalation is commonly used in catalysis, synthesis of main group complexes, and synthesis of transition metal complexes.
Types of transmetalation
There are two main types of transmetalation, redox-transmetalation (RT) and redox-transmetalation/ligand-exchange (RTLE). Below, M1 is usually a 4d or 5d transition metal and M2 is usually a main group or 3d transition metal. By looking at the electronegativities of the metals and ligands, one can predict whether the RT or RTLE reaction will proceed and what products the reaction will yield. For example, one can predict that the addition of 3 HgPh2 to 2 Al will yield 3 Hg and 2 AlPh3 because Hg is a more electronegative element than Al.
Redox-transmetalation
- M1n+–R + M2 → M1 + M2n+–R.
In redox-transmetalation a ligand is transferred from one metal to the other through an intermolecular mechanism. During the reaction one of the metal centers is oxidized and the other is reduced. The electronegativities of the metals and ligands is what causes the reaction to go forward. If M1 is more electronegative than M2, it is thermodynamically favorable for the R group to coordinate to the less electronegative M2.
Redox-transmetalation/ligand-exchange
- M1–R + M2–X → M1–X + M2–R.
In redox-transmetalation/ligand exchange the ligands of two metal complexes switch places with each other, bonding with the other metal center. The R ligand can be an alkyl, aryl, alkynyl, or allyl group and the X ligand can be a halogen, pseudo-halogen, alkyl, or aryl group. The reaction can proceed by two possible intermediate steps. The first is an associative intermediate, where the R and X ligands bridge the two metals, stabilizing the transition state. The second and less common intermediate is the formation of a cation where R is bridging the two metals and X is anionic. The RTLE reaction proceeds in a concerted manner. Like in RT reactions, the reaction is driven by electronegativity values. The X ligand is attracted to highly electropositive metals. If M1 is a more electropositive metal than M2, it is thermodynamically favorable for the exchange of the R and X ligands to occur.
Applications
Cross-coupling reactions
Transmetalation is often used as a step in the catalytic cycles of cross-coupling reactions. Some of the cross-coupling reactions that include a transmetalation step are Stille cross-coupling, Suzuki cross-coupling, Sonogashira cross-coupling, and Negishi cross-coupling. The most useful cross-coupling catalysts tend to be ones that contain palladium. Cross-coupling reactions have the general form of R′–X + M–R → R′–R + M–X and are used to form C–C bonds. R and R′ can be any carbon fragment. The identity of the metal, M, depends on which cross-coupling reaction is being used. Stille reactions use tin, Suzuki reactions use boron, Sonogashira reactions use copper, and Negishi reactions use zinc. The transmetalation step in palladium catalyzed reactions involve the addition of an R–M compound to produce an R′–Pd–R compound. Cross-coupling reactions have a wide range of applications in synthetic chemistry including the area of medicinal chemistry. The Stille reaction has been used to make an antitumor agent, (±)-epi-jatrophone;[3] the Suzuki reaction has been used to make an antitumor agent, oximidine II;[4] the Sonogashira reaction has been used to make an anticancer drug, eniluracil;[5] and the Negishi reaction has been used to make the carotenoid β-carotene via a transmetalation cascade.[6]
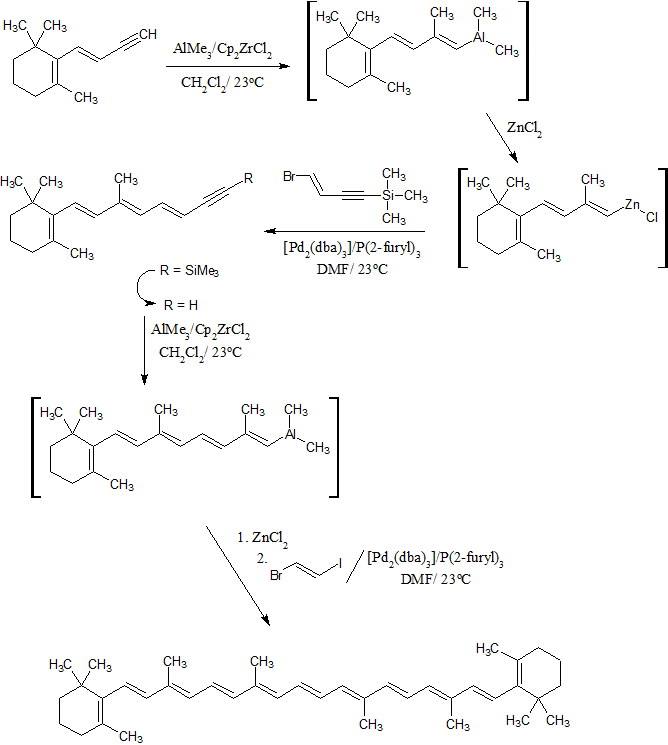 Figure 1. Synthesis of β-carotene by Negishi cross-coupling and transmetalation cascades.[1]
Figure 1. Synthesis of β-carotene by Negishi cross-coupling and transmetalation cascades.[1]
Lanthanides
Lanthanide organometallic complexes have been synthesized by RT and RTLE. Lanthanides are very electropositive elements.
Organomercurials, such as HgPh2, are common kinetically inert RT and RTLE reagents that allow functionalized derivatives to be synthesized, unlike organolithiums and Grignard reagents.[7] Diarylmercurials are often used to synthesize lanthanide organometallic complexes. Hg(C6F5)2 is a better RT reagent to use with lanthanides than HgPh2 because it does not require a step to activate the metal.[8] However, phenyl-substituted lanthanide complexes are more thermally stable than the pentafluorophenyl complexes. The use of HgPh2 led to the synthesis of a ytterbium complex with different oxidation states on the two Yb atoms:[9]
- Yb(C10H8)(THF)2 + HgPh2 → YbIIYbIIIPh5(THF)4
In the Ln(C6F5)2 complexes, where Ln = Yb, Eu, or Sm, the Ln–C bonds are very reactive, making them useful in RTLE reactions. Protic substrates have been used as a reactant with the Ln(C6F5)2 complex as shown: Ln(C6F5)2 + 2LH → Ln(L)2 + 2C6F5H. It is possible to avoid the challenges of working with the unstable Ln(C6F5)2 complex by forming it in situ by the following reaction:
- Ln + HgR2 + 2 LH → Ln(L)2 + Hg + 2 RH
Organotins are also kinetically inert RT and RTLE reagents that have been used in a variety of organometallic reactions. They have applications to the synthesis of lanthanide complexes, such as in the following reaction:[10]
- Yb + Sn(N(SiMe3)2)2 → Yb(N(SiMe3)2)2 + Sn
Actinides
RT can be used to synthesize actinide complexes. RT has been used to synthesize uranium halides using uranium metal and mercury halides as shown:
- U + HgX → UX + Hg (X = Cl, Br, I)[11]
This actinide RT reaction can be done with multiple mercury compounds to coordinate ligands other than halogens to the metal:
- 2 U + 3 (C5H5)2Hg + HgCl2 → 2 (C5H5)3UCl + 4 Hg
Alkaline earth metals
Alkaline earth metal complexes have been synthesized by RTLE, employing the same methodology used in synthesizing lanthanide complexes. The use of diphenylmercury in alkaline-earth metal reactions leads to the production of elemental mercury. The handling and disposal of elemental mercury is challenging due to its toxicity to humans and the environment. This led to the desire for an alternative RTLE reagent that would be less toxic and still very effective. Triphenylbismuth, BiPh3, was discovered to be a suitable alternative.[12] Mercury and bismuth have similar electronegativity values and behave similarly in RTLE reactions. BiPh3 has been used to synthesize alkaline-earth metal amides and alkaline-earth metal cyclopentadienides. The difference between HgPh2 and BiPh3 in these syntheses was that the reaction time was longer when using BiPh3.
References
- ↑ 1.0 1.1 Spessard, Gary O.; Miessler, Gary L. (2010). Organometallic Chemistry. New York, NY: Oxford University Press. ISBN 978-0195330991.
- ↑ Osakada, Kohtaro (2003). Fundamentals of Molecular Catalysis. Amsterdam: Elsevier. ISBN 0444509216. https://books.google.com/books?id=-5H55P2U-kYC.
- ↑ Gyorkos, Albert C.; Stille, John K.; Hegedus, Louis S. (1990). "The total synthesis of (±)-epi-jatrophone and (±)-jatrophone using palladium-catalyzed carbonylative coupling of vinyl triflates with vinyl stannanes as the macrocycle-forming step". J. Am. Chem. Soc. 112 (23): 8465–8472. doi:10.1021/ja00179a035.
- ↑ Molander, Gary A.; Dehmel, Florian (2004). "Formal Total Synthesis of Oximidine II via a Suzuki-Type Cross-Coupling Macrocyclization Employing Potassium Organotrifluoroborates". J. Am. Chem. Soc. 126 (33): 10313–10318. doi:10.1021/ja047190o. PMID 15315445.
- ↑ Cooke, Jason W. B.; Bright, Robert; Coleman, Mark J.; Jenkins, Kevin P. (2001). "Process Research and Development of a Dihydropyrimidine Dehydrogenase Inactivator: Large-Scale Preparation of Eniluracil Using a Sonogashira Coupling". Org. Process Res. Dev. 5 (4): 383–386. doi:10.1021/op0100100.
- ↑ Zeng, Fanxing; Negishi, Ei-Ichi (2001). "A Novel, Selective, and Efficient Route to Carotenoids and Related Natural Products via Zr-Catalyzed Carboalumination and Pd- and Zn-Catalyzed Cross Coupling". Org. Lett. 3 (5): 719–722. doi:10.1021/ol000384y. PMID 11259045.
- ↑ Vicente, José; Arcas, Aurelia; Gálvez López, María Dolores; Jones, Peter G. (2004). "Bis(2,6-dinitroaryl)platinum Complexes. 1. Synthesis through Transmetalation Reactions". Organometallics 23 (14): 3521–3527. doi:10.1021/om049801r.
- ↑ Deacon, Glen B.; Forsyth, Craig M.; Nickel, Siegbert (2002). "Bis(pentafluorophenyl)mercury—a versatile synthon in organo-, organooxo-, and organoamido-lanthanoid chemistry". J. Organomet. Chem. 647 (1–2): 50–60. doi:10.1016/S0022-328X(01)01433-4.
- ↑ Bochkarev, Mikhail N.; Khramenkov, Vladimir V.; Radkov, Yury F.; Zakharov, Lev N.; Struchkov, Yury T. (1992). "Synthesis and characterization of pentaphenyldiytterbium Ph2Yb(THF)(μ-Ph)3Yb(THF)3". J. Organomet. Chem. 429: 27–39. doi:10.1016/0022-328X(92)83316-A.
- ↑ Cetinkaya, Bekir; Hitchcock, Peter B.; Lappert, Michael F.; Smith, Richard G. (1992). "The first neutral, mononuclear 4f metal thiolates and new methods for corresponding aryl oxides and bis(trimethylsilyl)amides". J. Chem. Soc., Chem. Commun. 13 (13): 932–934. doi:10.1039/C39920000932.
- ↑ Deacon, G.B.; Tuong, T.D. (1988). "A simple redox transmetallation synthesis of uranium tetrachloride and related preparations of uranium triiodide and chlorotris(cyclopentadienyl)uranium(IV)". Polyhedron 7 (3): 249–250. doi:10.1016/S0277-5387(00)80561-6.
- ↑ Gillett-Kunnath, Miriam M.; MacLellan, Jonathan G.; Forsyth, Craig M.; Andrews, Philip C.; Deacon, Glen B.; Ruhlandt-Senge, Karin (2008). "BiPh3—A convenient synthon for heavy alkaline-earth metal amides". Chem. Commun. 37 (37): 4490–4492. doi:10.1039/b806948d. PMID 18802600.
 |  |


{kind=link}