Biology:Single cell sequencing
Single-cell sequencing examines the sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment.[1] For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types.[2] In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.[3]
Background
A typical human cell consists of about 2 x 3.3 billion base pairs of DNA and 600 million mRNA bases. Usually, a mix of millions of cells is used in sequencing the DNA or RNA using traditional methods like Sanger sequencing or Illumina sequencing. By deep sequencing of DNA and RNA from a single cell, cellular functions can be investigated extensively.[1] Like typical next-generation sequencing experiments, single-cell sequencing protocols generally contain the following steps: isolation of a single cell, nucleic acid extraction and amplification, sequencing library preparation, sequencing, and bioinformatic data analysis. It is more challenging to perform single-cell sequencing than sequencing from cells in bulk. The minimal amount of starting materials from a single cell makes degradation, sample loss, and contamination exert pronounced effects on the quality of sequencing data. In addition, due to the picogram level of the number of nucleic acids used,[4] heavy amplification is often needed during sample preparation of single-cell sequencing, resulting in uneven coverage, noise, and inaccurate quantification of sequencing data.
Recent technical improvements make single-cell sequencing a promising tool for approaching a set of seemingly inaccessible problems. For example, heterogeneous samples, rare cell types, cell lineage relationships, mosaicism of somatic tissues, analyses of microbes that cannot be cultured, and disease evolution can all be elucidated through single-cell sequencing.[5] Single-cell sequencing was selected as the method of the year 2013 by Nature Publishing Group.[6]
Genome (DNA) sequencing
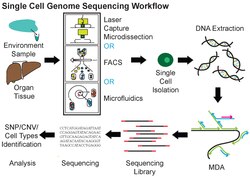
Single-cell DNA genome sequencing involves isolating a single cell, amplifying the whole genome or region of interest, constructing sequencing libraries, and then applying next-generation DNA sequencing (for example Illumina, Ion Torrent, MGI). Single-cell DNA sequencing has been widely applied in mammalian systems to study normal physiology and disease. Single-cell resolution can uncover the roles of genetic mosaicism or intra-tumor genetic heterogeneity in cancer development or treatment response.[7] In the context of microbiomes, a genome from a single unicellular organism is referred to as a single amplified genome (SAG). Advancements in single-cell DNA sequencing have enabled collecting of genomic data from uncultivated prokaryotic species present in complex microbiomes.[8] Although SAGs are characterized by low completeness and significant bias, recent computational advances have achieved the assembly of near-complete genomes from composite SAGs.[9] Data obtained from microorganisms might establish processes for culturing in the future.[10] Some of the genome assembly tools used in single cell single-cellencing include SPAdes, IDBA-UD, Cortex, and HyDA.[11]
Methods
A list of more than 100 different single-cell omics methods has been published.[12]
Multiple displacement amplification (MDA) is a widely used technique, enabling amplifying femtograms of DNA from bacterium to micrograms for sequencing. Reagents required for MDA reactions include: random primers and DNA polymerase from bacteriophage phi29. In 30 degree isothermal reaction, DNA is amplified with included reagents. As the polymerases manufacture new strands, a strand displacement reaction takes place, synthesizing multiple copies from each template DNA. At the same time, the strands that were extended antecedently will be displaced. MDA products result in a length of about 12 kb and ranges up to around 100 kb, enabling its use in DNA sequencing.[10] In 2017, a major improvement to this technique, called WGA-X, was introduced by taking advantage of a thermostable mutant of the phi29 polymerase, leading to better genome recovery from individual cells, in particular those with high G+C content.[13] MDA has also been implemented in a microfluidic droplet-based system to achieve a highly parallelized single-cell whole genome amplification. By encapsulating single-cells in droplets for DNA capture and amplification, this method offers reduced bias and enhanced throughput compared to conventional MDA.[14]
Another common method is MALBAC.[15] ThAs done in MDA, this method begins with isothermal amplificationbut the primers are flanked with a “common” sequence for downstream PCR amplification. As the preliminary amplicons are generated, the common sequence promotes self-ligation and the formation of “loops” to prevent further amplification. In contrast with MDA, the highly branched DNA network is not formed. Instead,, the loops are denatured in another temperature cycle allowing the fragments to be amplified with PCR. MALBAC has also been implemented in a microfluidic device, but the amplification performance was not significantly improved by encapsulation in nanoliter droplets.[16]
Comparing MDA and MALBAC, MDA results in better genome coverage, but MALBAC provides more even coverage across the genome. MDA could be more effective for identifying SNPs, whereas MALBAC is preferred for detecting copy number variants. While performing MDA with a microfluidic device markedly reduces bias and contamination, the chemistry involved in MALBAC does not demonstrate the same potential for improved efficiency.
A method particularly suitable for the discovery of genomic structural variation is Single-cell DNA template strand sequencing (a.k.a. Strand-seq).[17] Using the principle of single-cell tri-channel processing, which uses joint modelling of read-orientation, read-depth, and haplotype-phase, Strand-seq enables discovery of the full spectrum of somatic structural variation classes ≥200kb in size. Strand-seq overcomes limitations of whole genome amplification based methods for identification of somatic genetic variation classes in single cells,[18] because it is not susceptible against read chimers leading to calling artefacts (discussed in detail in the section below), and is less affected by drop outs. The choice of method depends on the goal of the sequencing because each method presents different advantages.[7]
Limitations
MDA of individual cell genomes results in highly uneven genome coverage, i.e. relative overrepresentation and underrepresentation of various regions of the template, leading to loss of some sequences. There are two components to this process: a) stochastic over- and under-amplification of random regions; and b) systematic bias against high %GC regions. The stochastic component may be addressed by pooling single-cell MDA reactions from the same cell type, by employing fluorescent in situ hybridization (FISH) and/or post-sequencing confirmation.[10] The bias of MDA against high %GC regions can be addressed by using thermostable polymerases, such as in the process called WGA-X.[13]
Single-nucleotide polymorphisms (SNPs), which are a big part of genetic variation in the human genome, and copy number variation (CNV), pose problems in single cell sequencing, as well as the limited amount of DNA extracted from a single cell. Due to scant amounts of DNA, accurate analysis of DNA poses problems even after amplification since coverage is low and is susceptible to errors. With MDA, average genome coverage is less than 80% and SNPs that are not covered by sequencing reads will be opted out. In addition, MDA shows a high ratio of allele dropout, not detecting alleles from heterozygous samples. Various SNP algorithms are currently in use but none are specific to single-cell sequencing. MDA with CNV also poses the problem of identifying false CNVs that conceal the real CNVs. To solve this, when patterns can be generated from false CNVs, algorithms can detect and eradicate this noise to produce true variants.[19]
Strand-seq overcomes limitations of methods based on whole genome amplification for genetic variant calling: Since Strand-seq does not require reads (or read pairs) transversing the boundaries (or breakpoints) of CNVs or copy-balanced structural variant classes, it is less susceptible to common artefacts of single-cell methods based on whole genome amplification, which include variant calling dropouts due to missing reads at the variant breakpoint and read chimera.[7][18]Strand-seq discovers the full spectrum of structural variation classes of at least 200kb in size, including breakage-fusion-bridge cycles and chromothripsis events, as well as balanced inversions, and copy-number balanced or imbalanced translocations.[18]" Structural variant calls made by Strand-seq are resolved by chromosome-length haplotype, which provides additional variant calling specificity.[18] As a current limitation, Strand-seq requires dividing cells for strand-specific labelling using bromodeoxyuridine (BrdU), and the method does not detect variants smaller than 200kb in size, such as mobile element insertions.
Applications
Microbiomes are among the main targets of single cell genomics due to the difficulty of culturing the majority of microorganisms in most environments. Single-cell genomics is a powerful way to obtain microbial genome sequences without cultivation. This approach has been widely applied on marine, soil, subsurface, organismal, and other types of microbiomes in order to address a wide array of questions related to microbial ecology, evolution, public health and biotechnology potential.[20][21][22][23][24][25][26][27][28]
Cancer sequencing is also an emerging application of scDNAseq. Fresh or frozen tumors may be analyzed and categorized with respect to SCNAs, SNVs, and rearrangements quite well using whole-genome DNAS approaches.[29] Cancer scDNAseq is particularly useful for examining the depth of complexity and compound mutations present in amplified therapeutic targets such as receptor tyrosine kinase genes (EGFR, PDGFRA etc.) where conventional population-level approaches of the bulk tumor are not able to resolve the co-occurrence patterns of these mutations within single cells of the tumor. Such overlap may provide redundancy of pathway activation and tumor cell resistance.
DNA methylome sequencing

Single-cell DNA methylome sequencing quantifies DNA methylation. There are several known types of methylation that occur in nature, including 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), 6-methyladenine (6mA), and 4mC 4-methylcytosine (4mC). In eukaryotes, especially animals, 5mC is widespread along the genome and plays an important role in regulating gene expression by repressing transposable elements.[31] Sequencing 5mC in individual cells can reveal how epigenetic changes across genetically identical cells from a single tissue or population give rise to cells with different phenotypes.
Methods
Bisulfite sequencing has become the gold standard in detecting and sequencing 5mC in single cells.[32] Treatment of DNA with bisulfite converts cytosine residues to uracil, but leaves 5-methylcytosine residues unaffected. Therefore, DNA that has been treated with bisulfite retains only methylated cytosines. To obtain the methylome readout, the bisulfite-treated sequence is aligned to an unmodified genome. Whole genome bisulfite sequencing was achieved in single cells in 2014.[33] The method overcomes the loss of DNA associated with the typical procedure, where sequencing adapters are added prior to bisulfite fragmentation. Instead, the adapters are added after the DNA is treated and fragmented with bisulfite, allowing all fragments to be amplified by PCR.[34] Using deep sequencing, this method captures ~40% of the total CpGs in each cell. With existing technology DNA cannot be amplified prior to bisulfite treatment, as the 5mC marks will not be copied by the polymerase.
Single-cell reduced representation bisulfite sequencing (scRRBS) is another method.[35] This method leverages the tendency of methylated cytosines to cluster at CpG islands (CGIs) to enrich for areas of the genome with a high CpG content. This reduces the cost of sequencing compared to whole-genome bisulfite sequencing, but limits the coverage of this method. When RRBS is applied to bulk samples, the majority of the CpG sites in gene promoters are detected, but site in gene promoters only account for 10% of CpG sites in the entire genome.[36] In single cells, 40% of the CpG sites from the bulk sample are detected. To increase coverage, this method can also be applied to a small pool of single cells. In a sample of 20 pooled single cells, 63% of the CpG sites from the bulk sample were detected. Pooling single cells is one strategy to increase methylome coverage, but at the cost of obscuring the heterogeneity in the population of cells.
Limitations
While bisulfite sequencing remains the most widely used approach for 5mC detection, the chemical treatment is harsh and fragments and degrades the DNA. This effect is exacerbated when moving from bulk samples to single cells. Other methods to detect DNA methylation include methylation-sensitive restriction enzymes. Restriction enzymes also enable the detection of other types of methylation, such as 6mA with DpnI.[37] Nanopore-based sequencing also offers a route for direct methylation sequencing without fragmentation or modification to the original DNA. Nanopore sequencing has been used to sequence the methylomes of bacteria, which are dominated by 6mA and 4mC (as opposed to 5mC in eukaryotes), but this technique has not yet been scaled down to single cells.[38]
Applications
Single-cell DNA methylation sequencing has been widely used to explore epigenetic differences in genetically similar cells. To validate these methods during their development, the single-cell methylome data of a mixed population were successfully classified by hierarchal clustering to identify distinct cell types.[35] Another application is studying single cells during the first few cell divisions in early development to understand how different cell types emerge from a single embryo.[39] Single-cell whole-genome bisulfite sequencing has also been used to study rare but highly active cell types in cancer such as circulating tumor cells (CTCs).[40]

Transposase-accessible chromatin sequencing (scATAC-seq)
Single cell transposase-accessible chromatin sequencing maps chromatin accessibility across the genome. A transposase inserts sequencing adapters directly into open regions of chromatin, allowing those regions to be amplified and sequenced.[41]
Transcriptome sequencing (scRNA-seq)
Standard methods such as microarrays and bulk RNA-seq analyze the RNA expression from large populations of cells. These measurements may obscure critical differences between individual cells in mixed-cell populations.[42][43]
Single-cell RNA sequencing (scRNA-seq) provides the expression profiles of individual cells and is considered the gold standard for defining cell states and phenotypes as of 2020.[44] Although it is impossible to obtain complete information on every RNA expressed by each cell, due to the small amount of material available, gene expression patterns can be identified through gene clustering analyses.[45] This can uncover rare cell types within a cell population that may never have been seen before. For example, one group of scientists performing scRNA-seq on neuroblastoma tumor tissue identified a rare pan-neuroblastoma cancer cell, which may be attractive for novel therapy approaches.[46]
Methods

Current scRNA-seq protocols involve isolating single cells and their RNA, and then following the same steps as bulk RNA-seq: reverse transcription (RT), amplification, library generation and sequencing. Early methods separated individual cells into separate wells; more recent methods encapsulate individual cells in droplets in a microfluidic device, where the reverse transcription reaction takes place, converting RNAs to cDNAs. Each droplet carries a DNA "barcode" that uniquely labels the cDNAs derived from a single cell. Once reverse transcription is complete, the cDNAs from many cells can be mixed together for sequencing, because transcripts from a particular cell are identified by the unique barcode.[47][48]
Challenges for scRNA-Seq include preserving the initial relative abundance of mRNA in a cell and identifying rare transcripts.[49] The reverse transcription step is critical as the efficiency of the RT reaction determines how much of the cell’s RNA population will be eventually analyzed by the sequencer. The processivity of reverse transcriptases and the priming strategies used may affect full-length cDNA production and the generation of libraries biased toward 3’ or 5' end of genes.
In the amplification step, either PCR or in vitro transcription (IVT) is currently used to amplify cDNA. One of the advantages of PCR-based methods is the ability to generate full-length cDNA. However, different PCR efficiency on particular sequences (for instance, GC content and snapback structure) may also be exponentially amplified, producing libraries with uneven coverage. On the other hand, while libraries generated by IVT can avoid PCR-induced sequence bias, specific sequences may be transcribed inefficiently, thus causing sequence drop-out or generating incomplete sequences.[1][42] Several scRNA-seq protocols have been published: Tang et al.,[50] STRT,[51] SMART-seq,[52] SORT-seq,[53] CEL-seq,[54] RAGE-seq,[55] Quartz-seq.[56] , and C1-CAGE.[57] These protocols differ in terms of strategies for reverse transcription, cDNA synthesis and amplification, and the possibility to accommodate sequence-specific barcodes (i.e., UMIs) or the ability to process pooled samples.[58]
In 2017, two approaches were introduced to simultaneously measure single-cell mRNA and protein expression through oligonucleotide-labeled antibodies known as REAP-seq,[59] and CITE-seq.[60] Collecting cellular contents following electrophysiological recording using patch-clamp has also allowed development of the Patch-Seq method, which is steadily gaining ground in neuroscience.[61]
Example of a droplet based platform - 10X method
This platform of single cell RNA sequencing allows to analyze transcriptomes on a cell-by-cell basis by the use of microfluidic partitioning to capture single cells and prepare next-generation sequencing (NGS) cDNA libraries.[62] The droplets based platform enables massively parallel sequencing of mRNA in a large numbers of individual cells by capturing single cell in oil droplet.[63]
Overall, in a first stage individual cells are captured separately and lysed, then reverse transcription (RT) of mRNA is performed and cDNA library is obtained. To select mRNA, the RT is performed with a single-stranded sequence of deoxythymine (oligo dT) primer which bind specifically the poly(A) tail of mRNA molecules. Subsequently, the amplified cDNA library is used for sequencing.[64]
So, the first step of the method is the single cell encapsulation and library preparation. Cells are encapsulated into Gel Beads-in-emulsion (GEMs) thanks to an automate. To form these vesicle, the automate uses a microfluidic chip and combines all components with oil. Each functional GEM contains a single cell, a single Gel Bead, and RT reagents. On the Gel Bead, olignonucleotides composed by 4 distincts parts are bind: PCR primer (essencial for the sequencing) ; 10X barcoded oligonucleotides ; Unique Molecular Identifier (UMI) sequence ; PolydT sequence (that enables capture of poly-adenylated mRNA molecules).[65] Within each GEM reaction vesicle, a single cell is lysed and undergo reverse transcription. cDNA from the same cell are identified thanks to a common 10X barcode. In addition, the number of UMIs express the gene expression level and its analyse allows to detect highly variable genes. Those data are often used for either cellular phenotype classification or new subpopulation identification.[66]
The final step of the plateform is the sequencing. Libraries generated can be directly used for single cell whole transcriptome sequencing or target sequencing workflows. The sequencing is performed by using the Illumina dye sequencing method. This sequencing method is based on sequencing by synthesis (SBS) principle and the use of reversible dye-terminator that enables the identification of each single nucleotid. In order to read the transcript sequences on one end, and the barcode and UMI on the other end, paired-end sequencing readers are required.[67]
The droplet-based platform allows to detect rare cell type thanks to its hight throuhput. In fact, 500 to 10,000 cells are carptured per sample from a single cell suspension. The protocol is performed easily and allows a high cell recovery rate of up to 65%. The global workflow of the droplet-based platform takes 8 hours and so is faster than the Microwell-based method (BD Rhapsody), which takes 10 hours. However, it presents some limitations as the need of fresh samples and the final detection of only 10% mRNA.
The major difference between the droplet-based method and the microwell-based method is the technique used for partitioning cells.[68]
Limitations
Most RNA-seq methods depend on poly(A) tail capture to enrich mRNA and deplete abundant and uninformative rRNA. Thus, they are often restricted to sequencing polyadenylated mRNA molecules. However, recent studies are now starting to appreciate the importance of non-poly(A) RNA, such as long-noncoding RNA and microRNAs in gene expression regulation. Small-seq is a single-cell method that captures small RNAs (<300 nucleotides) such as microRNAs, fragments of tRNAs and small nucleolar RNAs in mammalian cells.[69] This method uses a combination of “oligonucleotide masks” (that inhibit the capture of highly abundant 5.8S rRNA molecules) and size selection to exclude large RNA species such as other highly abundant rRNA molecules. To target larger non-poly(A) RNAs, such as long non-coding mRNA, histone mRNA, circular RNA, and enhancer RNA, size selection is not applicable for depleting the highly abundant ribosomal RNA molecules (18S and 28s rRNA).[70] Single-cell RamDA-Seq is a method that achieves this by performing reverse transcription with random priming (random displacement amplification) in the presence of “not so random” (NSR) primers specifically designed to avoid priming on rRNA molecule.[71] While this method successfully captures full-length total RNA transcripts for sequencing and detected a variety of non-poly(A) RNAs with high sensitivity, it has some limitations. The NSR primers were carefully designed according to rRNA sequences in the specific organism (mouse), and designing new primer sets for other species would take considerable effort. Recently, a CRISPR-based method named scDASH (single-cell depletion of abundant sequences by hybridization) demonstrated another approach to depleting rRNA sequences from single-cell total RNA-seq libraries.[72]
Bacteria and other prokaryotes are currently not amenable to single-cell RNA-seq due to the lack of polyadenylated mRNA. Thus, the development of single-cell RNA-seq methods that do not depend on poly(A) tail capture will also be instrumental in enabling single-cell resolution microbiome studies. Bulk bacterial studies typically apply general rRNA depletion to overcome the lack of polyadenylated mRNA on bacteria, but at the single-cell level, the total RNA found in one cell is too small.[70] Lack of polyadenylated mRNA and scarcity of total RNA found in single bacteria cells are two important barriers limiting the deployment of scRNA-seq in bacteria.
Applications
scRNA-Seq is becoming widely used across biological disciplines including Developmental biology,[73] Neurology,[74] Oncology,[75][76][77] Immunology,[78][79] Cardiovascular research[80][81] and Infectious disease.[82][83]
Using machine learning methods, data from bulk RNA-Seq has been used to increase the signal/noise ratio in scRNA-Seq. Specifically, scientists have used gene expression profiles from pan-cancer datasets in order to build coexpression networks, and then have applied these on single cell gene expression profiles, obtaining a more robust method to detect the presence of mutations in individual cells using transcript levels.[84]
Some scRNA-seq methods have also been applied to single cell microorganisms. SMART-seq2 has been used to analyze single cell eukaryotic microbes, but since it relies on poly(A) tail capture, it has not been applied in prokaryotic cells.[85] Microfluidic approaches such as Drop-seq and the Fluidigm IFC-C1 devices have been used to sequence single malaria parasites or single yeast cells.[86][87] The single-cell yeast study sought to characterize the heterogeneous stress tolerance in isogenic yeast cells before and after the yeast are exposed to salt stress. Single-cell analysis of the several transcription factors by scRNA-seq revealed heterogeneity across the population. These results suggest that regulation varies among members of a population to increase the chances of survival for a fraction of the population.
The first single-cell transcriptome analysis in a prokaryotic species was accomplished using the terminator exonuclease enzyme to selectively degrade rRNA and rolling circle amplification (RCA) of mRNA.[88] In this method, the ends of single-stranded DNA were ligated together to form a circle, and the resulting loop was then used as a template for linear RNA amplification. The final product library was then analyzed by microarray, with low bias and good coverage. However, RCA has not been tested with RNA-seq, which typically employs next-generation sequencing. Single-cell RNA-seq for bacteria would be highly useful for studying microbiomes. It would address issues encountered in conventional bulk metatranscriptomics approaches, such as failing to capture species present in low abundance, and failing to resolve heterogeneity among cell populations.
scRNA-Seq has provided considerable insight into the development of embryos and organisms, including the worm Caenorhabditis elegans,[89] and the regenerative planarian Schmidtea mediterranea[90][91] and axolotl Ambystoma mexicanum.[92][93] The first vertebrate animals to be mapped in this way were Zebrafish[94][95][96] and Xenopus laevis.[97] In each case multiple stages of the embryo were studied, allowing the entire process of development to be mapped on a cell-by-cell basis. Science recognized these advances as the 2018 Breakthrough of the Year.[98]
A molecular cell atlas of mice testes was established to define BDE47-induced prepubertal testicular toxicity using the ScRNA-seq approach, providing novel insight into our understanding of the underlying mechanisms and pathways involved in BDE47-associated testicular injury at a single-cell resolution.[99]
Considerations
Isolation of single cells
There are several ways to isolate individual cells prior to whole genome amplification and sequencing. Fluorescence-activated cell sorting (FACS) is a widely used approach. Individual cells can also be collected by micromanipulation, for example by serial dilution or by using a patch pipette or nanotube to harvest a single cell.[15][100] The advantages of micromanipulation are ease and low cost, but they are laborious and susceptible to misidentification of cell types under microscope. Laser-capture microdissection (LCM) can also be used for collecting single cells. Although LCM preserves the knowledge of the spatial location of a sampled cell within a tissue, it is hard to capture a whole single cell without also collecting the materials from neighboring cells.[42][101][102] High-throughput methods for single cell isolation also include microfluidics. Both FACS and microfluidics are accurate, automatic and capable of isolating unbiased samples. However, both methods require detaching cells from their microenvironments first, thereby causing perturbation to the transcriptional profiles in RNA expression analysis.[103][104]
Number of cells to be analyzed
scRNA-Seq
Generally speaking, for a typical bulk cell RNA-sequencing (RNA-seq) experiment, ten million reads are generated and a gene with higher than the threshold of 50 reads per kb per million reads (RPKM) is considered expressed. For a gene that is 1kb long, this corresponds to 500 reads and a minimum coefficient of variation (CV) of 4% under the assumption of the Poisson distribution. For a typical mammalian cell containing 200,000 mRNA, sequencing data from at least 50 single cells need to be pooled in order to achieve this minimum CV value. However, due to the efficiency of reverse transcription and other noise introduced in the experiments, more cells are required for accurate expression analyses and cell-type identification.[42]
See also
- Single-cell analysis
- Single-cell transcriptomics
- Single cell epigenomics
- DNA sequencing
- Whole genome sequencing
References
- ↑ 1.0 1.1 1.2 "The promise of single-cell sequencing". Nature Methods 11 (1): 25–27. January 2014. doi:10.1038/nmeth.2769. PMID 24524134.
- ↑ "Chronicling embryos, cell by cell, gene by gene". Science 360 (6387): 367. April 2018. doi:10.1126/science.360.6387.367. PMID 29700246. Bibcode: 2018Sci...360..367P.
- ↑ "Single-cell RNA-seq: advances and future challenges". Nucleic Acids Research 42 (14): 8845–8860. August 2014. doi:10.1093/nar/gku555. PMID 25053837.
- ↑ "On-chip separation and analysis of RNA and DNA from single cells". Analytical Chemistry 86 (4): 1953–1957. February 2014. doi:10.1021/ac4040218. PMID 24499009.
- ↑ "Single-cell sequencing". Nature Methods 11 (1): 18. January 2014. doi:10.1038/nmeth.2771. PMID 24524131.
- ↑ "Method of the year 2013". Nature Methods 11 (1): 1. January 2014. doi:10.1038/nmeth.2801. PMID 24524124.
- ↑ 7.0 7.1 7.2 "Single-cell genome sequencing: current state of the science". Nature Reviews. Genetics 17 (3): 175–188. March 2016. doi:10.1038/nrg.2015.16. PMID 26806412.
- ↑ "Genomes from uncultivated prokaryotes: a comparison of metagenome-assembled and single-amplified genomes". Microbiome 6 (1): 173. September 2018. doi:10.1186/s40168-018-0550-0. PMID 30266101.
- ↑ "Obtaining high-quality draft genomes from uncultured microbes by cleaning and co-assembly of single-cell amplified genomes". Scientific Reports 8 (1): 2059. February 2018. doi:10.1038/s41598-018-20384-3. PMID 29391438. Bibcode: 2018NatSR...8.2059K.
- ↑ 10.0 10.1 10.2 ""Single-cell genomic sequencing using Multiple Displacement Amplification". Current Opinion in Microbiology 10 (5): 510–516. October 2007. doi:10.1016/j.mib.2007.08.005. PMID 17923430."
- ↑ "Distilled single-cell genome sequencing and de novo assembly for sparse microbial communities". Bioinformatics 29 (19): 2395–2401. October 2013. doi:10.1093/bioinformatics/btt420. PMID 23918251. Bibcode: 2013arXiv1305.0062T.
- ↑ "Single-Cell-Omics.v2.3.13 @albertvilella" (in en-US). https://docs.google.com/spreadsheets/d/1IPe2ozb1Mny8sLvJaSE57RJr3oruiBoSudAVhSH-O8M/edit?usp=embed_facebook.
- ↑ 13.0 13.1 "Improved genome recovery and integrated cell-size analyses of individual uncultured microbial cells and viral particles". Nature Communications 8 (1): 84. July 2017. doi:10.1038/s41467-017-00128-z. PMID 28729688. Bibcode: 2017NatCo...8...84S.
- ↑ "Massively parallel whole genome amplification for single-cell sequencing using droplet microfluidics". Scientific Reports 7 (1): 5199. July 2017. doi:10.1038/s41598-017-05436-4. PMID 28701744. Bibcode: 2017NatSR...7.5199H.
- ↑ 15.0 15.1 "Genome-wide detection of single-nucleotide and copy-number variations of a single human cell". Science 338 (6114): 1622–1626. December 2012. doi:10.1126/science.1229164. PMID 23258894. Bibcode: 2012Sci...338.1622Z.
- ↑ "Microfluidic whole genome amplification device for single cell sequencing". Analytical Chemistry 86 (19): 9386–9390. October 2014. doi:10.1021/ac5032176. PMID 25233049.
- ↑ "DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution". Nature Methods 9 (11): 1107–1112. November 2012. doi:10.1038/nmeth.2206. PMID 23042453.
- ↑ 18.0 18.1 18.2 18.3 ""Single-cell analysis of structural variations and complex rearrangements with tri-channel processing". Nature Biotechnology 38 (3): 343–354. March 2020. doi:10.1038/s41587-019-0366-x. PMID 31873213."
- ↑ ""Current challenges in the bioinformatics of single cell genomics". Frontiers in Oncology 4 (7): 7. 2014. doi:10.3389/fonc.2014.00007. PMID 24478987."
- ↑ "Dissecting genomic diversity, one cell at a time". Nature Methods 11 (1): 19–21. January 2014. doi:10.1038/nmeth.2783. PMID 24524132.
- ↑ "Sequencing genomes from single cells by polymerase cloning". Nature Biotechnology 24 (6): 680–686. June 2006. doi:10.1038/nbt1214. PMID 16732271.
- ↑ "Single-cell genomics reveals organismal interactions in uncultivated marine protists". Science 332 (6030): 714–717. May 2011. doi:10.1126/science.1203163. PMID 21551060. Bibcode: 2011Sci...332..714Y.
- ↑ "Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean". Science 333 (6047): 1296–1300. September 2011. doi:10.1126/science.1203690. PMID 21885783. Bibcode: 2011Sci...333.1296S.
- ↑ "Assembling the marine metagenome, one cell at a time". PLOS ONE 4 (4): e5299. 2009-04-23. doi:10.1371/journal.pone.0005299. PMID 19390573. Bibcode: 2009PLoSO...4.5299W.
- ↑ "Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean". Proceedings of the National Academy of Sciences of the United States of America 110 (28): 11463–11468. July 2013. doi:10.1073/pnas.1304246110. PMID 23801761. Bibcode: 2013PNAS..11011463S.
- ↑ "Insights into the phylogeny and coding potential of microbial dark matter". Nature 499 (7459): 431–437. July 2013. doi:10.1038/nature12352. PMID 23851394. Bibcode: 2013Natur.499..431R.
- ↑ "Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus". Science 344 (6182): 416–420. April 2014. doi:10.1126/science.1248575. PMID 24763590. Bibcode: 2014Sci...344..416K.
- ↑ "Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation". Science 358 (6366): 1046–1051. November 2017. doi:10.1126/science.aan8260. PMID 29170234. Bibcode: 2017Sci...358.1046P.
- ↑ "EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing". Cancer Discovery 4 (8): 956–971. August 2014. doi:10.1158/2159-8290.CD-13-0879. PMID 24893890.
- ↑ "Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics". Cell Reports 10 (8): 1386–1397. March 2015. doi:10.1016/j.celrep.2015.02.001. PMID 25732828.
- ↑ "Genome-wide evolutionary analysis of eukaryotic DNA methylation". Science 328 (5980): 916–919. May 2010. doi:10.1126/science.1186366. PMID 20395474. Bibcode: 2010Sci...328..916Z.
- ↑ "A comparison of existing global DNA methylation assays to low-coverage whole-genome bisulfite sequencing for epidemiological studies". Epigenetics 12 (3): 206–214. March 2017. doi:10.1080/15592294.2016.1276680. PMID 28055307.
- ↑ "Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity". Nature Methods 11 (8): 817–820. August 2014. doi:10.1038/nmeth.3035. PMID 25042786.
- ↑ "Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging". Nucleic Acids Research 40 (17): e136. September 2012. doi:10.1093/nar/gks454. PMID 22649061.
- ↑ 35.0 35.1 "Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing". Genome Research 23 (12): 2126–2135. December 2013. doi:10.1101/gr.161679.113. PMID 24179143.
- ↑ "Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling". Nature Protocols 6 (4): 468–481. April 2011. doi:10.1038/nprot.2010.190. PMID 21412275.
- ↑ "N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas". Cell 161 (4): 879–892. May 2015. doi:10.1016/j.cell.2015.04.010. PMID 25936837.
- ↑ "Deciphering bacterial epigenomes using modern sequencing technologies". Nature Reviews. Genetics 20 (3): 157–172. March 2019. doi:10.1038/s41576-018-0081-3. PMID 30546107.
- ↑ "The DNA methylation landscape of human early embryos". Nature 511 (7511): 606–610. July 2014. doi:10.1038/nature13544. PMID 25079557. Bibcode: 2014Natur.511..606G.
- ↑ "Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding". Cell 176 (1–2): 98–112.e14. January 2019. doi:10.1016/j.cell.2018.11.046. PMID 30633912.
- ↑ "Single-Cell Sequencing Sifts through Multiple Omics". 1 Jul 2019. https://www.genengnews.com/topics/omics/single-cell-sequencing-sifts-through-multiple-omics/.
- ↑ 42.0 42.1 42.2 42.3 ""Single-cell sequencing-based technologies will revolutionize whole-organism science". Nature Reviews. Genetics 14 (9): 618–630. September 2013. doi:10.1038/nrg3542. PMID 23897237."
- ↑ "The technology and biology of single-cell RNA sequencing". Molecular Cell 58 (4): 610–620. May 2015. doi:10.1016/j.molcel.2015.04.005. PMID 26000846.
- ↑ "Investigating Tumor Heterogeneity in Mouse Models". Annual Review of Cancer Biology 4 (1): 99–119. March 2020. doi:10.1146/annurev-cancerbio-030419-033413. PMID 34164589.
- ↑ Harris C (2020). Single Cell Transcriptome Analysis in Prostate Cancer (MSc). University of Otago. hdl:10523/10111.
- ↑ "Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell". Science Advances 7 (6): eabd3311. February 2021. doi:10.1126/sciadv.abd3311. PMID 33547074. Bibcode: 2021SciA....7.3311K.
- ↑ "Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells". Cell 161 (5): 1187–1201. May 2015. doi:10.1016/j.cell.2015.04.044. PMID 26000487.
- ↑ "Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets". Cell 161 (5): 1202–1214. May 2015. doi:10.1016/j.cell.2015.05.002. PMID 26000488.
- ↑ ""Methods, Challenges and Potentials of Single Cell RNA-seq". Biology 1 (3): 658–667. November 2012. doi:10.3390/biology1030658. PMID 24832513."
- ↑ "mRNA-Seq whole-transcriptome analysis of a single cell". Nature Methods 6 (5): 377–382. May 2009. doi:10.1038/NMETH.1315. PMID 19349980.
- ↑ "Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq". Genome Research 21 (7): 1160–1167. July 2011. doi:10.1101/gr.110882.110. PMID 21543516.
- ↑ "Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells". Nature Biotechnology 30 (8): 777–782. August 2012. doi:10.1038/nbt.2282. PMID 22820318.
- ↑ "A Single-Cell Transcriptome Atlas of the Human Pancreas". Cell Systems 3 (4): 385–394.e3. October 2016. doi:10.1016/j.cels.2016.09.002. PMID 27693023.
- ↑ "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification". Cell Reports 2 (3): 666–673. September 2012. doi:10.1016/j.celrep.2012.08.003. PMID 22939981.
- ↑ "High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes". Nature Communications 10 (1): 3120. July 2019. doi:10.1038/s41467-019-11049-4. PMID 31311926. Bibcode: 2019NatCo..10.3120S.
- ↑ "Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity". Genome Biology 14 (4): R31. April 2013. doi:10.1186/gb-2013-14-4-r31. PMID 23594475.
- ↑ "C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution". Nature Communications 10 (1): 360. January 2019. doi:10.1038/s41467-018-08126-5. PMID 30664627. Bibcode: 2019NatCo..10..360K.
- ↑ "How to design a single-cell RNA-sequencing experiment: pitfalls, challenges and perspectives". Briefings in Bioinformatics 20 (4): 1384–1394. July 2019. doi:10.1093/bib/bby007. PMID 29394315.
- ↑ "Multiplexed quantification of proteins and transcripts in single cells". Nature Biotechnology 35 (10): 936–939. October 2017. doi:10.1038/nbt.3973. PMID 28854175.
- ↑ "Simultaneous epitope and transcriptome measurement in single cells". Nature Methods 14 (9): 865–868. September 2017. doi:10.1038/nmeth.4380. PMID 28759029.
- ↑ "Assessing Transcriptome Quality in Patch-Seq Datasets". Frontiers in Molecular Neuroscience 11: 363. 2018-10-08. doi:10.3389/fnmol.2018.00363. PMID 30349457.
- ↑ Clark, Sheila. "Single cell RNA-seq: An introductory overview and tools for getting started". https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started.
- ↑ Gudapati, H; Dey, M; Ozbolat, I (September 2016). "A comprehensive review on droplet-based bioprinting: Past, present and future.". Biomaterials 102: 20–42. doi:10.1016/j.biomaterials.2016.06.012. PMID 27318933.
- ↑ Gao, C; Zhang, M; Chen, L (December 2020). "The Comparison of Two Single-cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium.". Current Genomics 21 (8): 602–609. doi:10.2174/1389202921999200625220812. PMID 33414681.
- ↑ "Chromium Single Cell Gene Expression Solution with Feature Barcoding technology". https://elixir-iib-training.github.io/2019-05-07-pozzuoli-singlecell/pres/Sadet_v3_with_Feature_Barcoding_RevA.pdf.
- ↑ Wang, X; He, Y; Zhang, Q; Ren, X; Zhang, Z (April 2021). "Direct Comparative Analyses of 10X Genomics Chromium and Smart-seq2.". Genomics, Proteomics & Bioinformatics 19 (2): 253–266. doi:10.1016/j.gpb.2020.02.005. PMID 33662621.
- ↑ KWOK, Hin; LUI, Schwan. "Single Cell (10X Genomics)". https://cpos.hku.hk/portfolio-item/single-cell-10x-genomics/.
- ↑ Gao, C; Zhang, M; Chen, L (December 2020). "The Comparison of Two Single-cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium.". Current Genomics 21 (8): 602–609. doi:10.2174/1389202921999200625220812. PMID 33414681.
- ↑ "Small-seq for single-cell small-RNA sequencing". Nature Protocols 13 (10): 2407–2424. October 2018. doi:10.1038/s41596-018-0049-y. PMID 30250291.
- ↑ 70.0 70.1 "Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs". Nature Communications 9 (1): 619. February 2018. doi:10.1038/s41467-018-02866-0. PMID 29434199. Bibcode: 2018NatCo...9..619H.
- ↑ "Digital transcriptome profiling using selective hexamer priming for cDNA synthesis". Nature Methods 6 (9): 647–649. September 2009. doi:10.1038/nmeth.1360. PMID 19668204.
- ↑ "Effective ribosomal RNA depletion for single-cell total RNA-seq by scDASH". PeerJ 9: e10717. 2021-01-15. doi:10.7717/peerj.10717. PMID 33520469.
- ↑ "Using single-cell genomics to understand developmental processes and cell fate decisions". Molecular Systems Biology 14 (4): e8046. April 2018. doi:10.15252/msb.20178046. PMID 29661792.
- ↑ "Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain". Nature Biotechnology 36 (5): 442–450. June 2018. doi:10.1038/nbt.4103. PMID 29608178.
- ↑ "Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience". Annals of Oncology 20 (1): 27–33. January 2009. doi:10.1093/annonc/mdn544. PMID 18695026.
- ↑ "Single-Cell Transcriptomic Analysis of Tumor Heterogeneity". Trends in Cancer 4 (4): 264–268. April 2018. doi:10.1016/j.trecan.2018.02.003. PMID 29606308.
- ↑ "A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade". Cell 175 (4): 984–997.e24. November 2018. doi:10.1016/j.cell.2018.09.006. PMID 30388455.
- ↑ "Single-Cell Genomics: Approaches and Utility in Immunology". Trends in Immunology 38 (2): 140–149. February 2017. doi:10.1016/j.it.2016.12.001. PMID 28094102.
- ↑ "Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation". Nature Communications 9 (1): 791. February 2018. doi:10.1038/s41467-017-02659-x. PMID 29476078. Bibcode: 2018NatCo...9..791S.
- ↑ "Decoding myofibroblast origins in human kidney fibrosis". Nature 589 (7841): 281–286. January 2021. doi:10.1038/s41586-020-2941-1. PMID 33176333. Bibcode: 2021Natur.589..281K.
- ↑ "Spatial multi-omic map of human myocardial infarction" (in en). Nature 608 (7924): 766–777. 2022-08-10. doi:10.1038/s41586-022-05060-x. ISSN 1476-4687. PMID 35948637. Bibcode: 2022Natur.608..766K.
- ↑ "Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses". Cell 162 (6): 1309–1321. September 2015. doi:10.1016/j.cell.2015.08.027. PMID 26343579.
- ↑ "Predicting bacterial infection outcomes using single cell RNA-sequencing analysis of human immune cells". Nature Communications 10 (1): 3266. July 2019. doi:10.1038/s41467-019-11257-y. PMID 31332193. Bibcode: 2019NatCo..10.3266B.
- ↑ "Pan-Cancer and Single-Cell Modeling of Genomic Alterations Through Gene Expression". Frontiers in Genetics 10: 671. 2019. doi:10.3389/fgene.2019.00671. PMID 31379928.
- ↑ "Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites". eLife 7: e33105. March 2018. doi:10.7554/eLife.33105. PMID 29580379.
- ↑ "Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites". Nature 551 (7678): 95–99. November 2017. doi:10.1038/nature24280. PMID 29094698. Bibcode: 2017Natur.551...95P.
- ↑ "Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress". PLOS Biology 15 (12): e2004050. December 2017. doi:10.1371/journal.pbio.2004050. PMID 29240790.
- ↑ "Transcript amplification from single bacterium for transcriptome analysis". Genome Research 21 (6): 925–935. June 2011. doi:10.1101/gr.116103.110. PMID 21536723.
- ↑ "Comprehensive single-cell transcriptional profiling of a multicellular organism". Science 357 (6352): 661–667. August 2017. doi:10.1126/science.aam8940. PMID 28818938. Bibcode: 2017Sci...357..661C.
- ↑ "Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics". Science 360 (6391): eaaq1723. May 2018. doi:10.1126/science.aaq1723. PMID 29674432.
- ↑ "Cell type transcriptome atlas for the planarian Schmidtea mediterranea". Science 360 (6391): eaaq1736. May 2018. doi:10.1126/science.aaq1736. PMID 29674431.
- ↑ "Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration". Science 362 (6413): eaaq0681. October 2018. doi:10.1126/science.aaq0681. PMID 30262634. Bibcode: 2018Sci...362..681G.
- ↑ "Transcriptomic landscape of the blastema niche in regenerating adult axolotl limbs at single-cell resolution". Nature Communications 9 (1): 5153. December 2018. doi:10.1038/s41467-018-07604-0. PMID 30514844. Bibcode: 2018NatCo...9.5153L.
- ↑ "Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo". Science 360 (6392): 981–987. June 2018. doi:10.1126/science.aar4362. PMID 29700229. Bibcode: 2018Sci...360..981W.
- ↑ "Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis". Science 360 (6392): eaar3131. June 2018. doi:10.1126/science.aar3131. PMID 29700225.
- ↑ "Single-cell lineage tracing by integrating CRISPR-Cas9 mutations with transcriptomic data". Nature Communications 11 (1): 3055. June 2020. doi:10.1038/s41467-020-16821-5. PMID 32546686. Bibcode: 2020NatCo..11.3055Z.
- ↑ "The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution". Science 360 (6392): eaar5780. June 2018. doi:10.1126/science.aar5780. PMID 29700227.
- ↑ "Science's 2018 Breakthrough of the Year: tracking development cell by cell". Science Magazine. American Association for the Advancement of Science. https://vis.sciencemag.org/breakthrough2018/finalists/.
- ↑ "Characterization of 2,2',4,4'-tetrabromodiphenyl ether (BDE47)-induced testicular toxicity via single-cell RNA-sequencing". Precision Clinical Medicine 5 (3): pbac016. September 2022. doi:10.1093/pcmedi/pbac016. PMID 35875604.
- ↑ "Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis". Nature Protocols 2 (3): 739–752. 2007. doi:10.1038/nprot.2007.79. PMID 17406636.
- ↑ "Distant metastasis occurs late during the genetic evolution of pancreatic cancer". Nature 467 (7319): 1114–1117. October 2010. doi:10.1038/nature09515. PMID 20981102. Bibcode: 2010Natur.467.1114Y.
- ↑ "Amplification of multiple genomic loci from single cells isolated by laser micro-dissection of tissues". BMC Biotechnology 8 (17): 17. February 2008. doi:10.1186/1472-6750-8-17. PMID 18284708.
- ↑ "Single-cell dissection of transcriptional heterogeneity in human colon tumors". Nature Biotechnology 29 (12): 1120–1127. November 2011. doi:10.1038/nbt.2038. PMID 22081019.
- ↑ "High-throughput microfluidic single-cell RT-qPCR". Proceedings of the National Academy of Sciences of the United States of America 108 (34): 13999–14004. August 2011. doi:10.1073/pnas.1019446108. PMID 21808033. Bibcode: 2011PNAS..10813999W.
External links
 |
