Biology:Glucocerebrosidase
 Generic protein structure example |
β-Glucocerebrosidase (also called acid β-glucosidase, D-glucosyl-N-acylsphingosine glucohydrolase, or GCase) is an enzyme with glucosylceramidase activity (EC 3.2.1.45) that cleaves by hydrolysis the β-glycosidic linkage of the chemical glucocerebroside, an intermediate in glycolipid metabolism that is abundant in cell membranes (particularly skin cells).[1] It is localized in the lysosome, where it remains associated with the lysosomal membrane.[2] β-Glucocerebrosidase is 497 amino acids in length and has a molecular mass of 59,700 Da.[citation needed]
Structure
β-Glucocerebrosidase is a member of the glycoside hydrolase family 30 and consists of three distinct domains (I-III).[3]
-
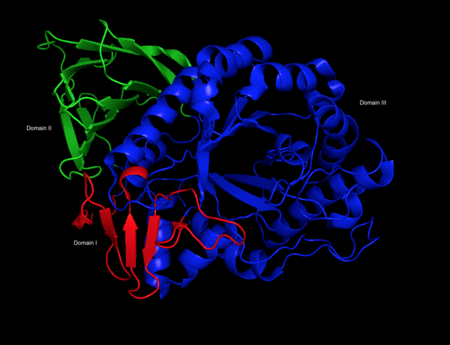 Three-dimensional PyMol rendering of glucocerebrosidase with three domains highlighted.
Three-dimensional PyMol rendering of glucocerebrosidase with three domains highlighted. -
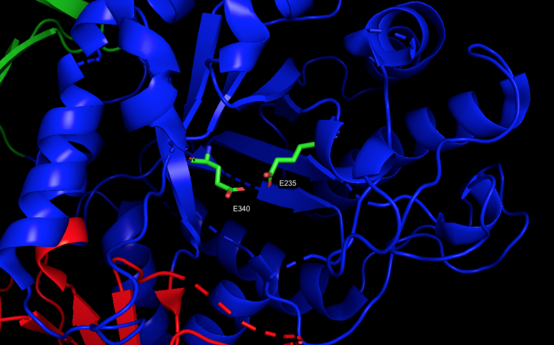 Three-dimensional PyMol rendering of glucocerebrosidase with catalytic residues highlighted.
Three-dimensional PyMol rendering of glucocerebrosidase with catalytic residues highlighted.


Domain I (residues 1–27 and 383–414) forms a three-stranded anti-parallel β-sheet. This domain contains two disulfide bridges that are necessary for correct folding, as well as a glycosylated residue (Asn19) that is required for catalytic activity in vivo. Domain II (residues 30–75 and 431–497) consists of two β-sheets that resemble an immunoglobulin fold. Domain III (residues 76–381 and 416–430) is homologous to a TIM barrel and is a highly conserved domain among glycoside hydrolases.[4] Domain III harbors the active site, which binds the substrate glucocerebroside in close proximity to the catalytic residues E340 and E235. Domains I and III are tightly associated, while domains II and III are joined by a disordered linker.[3]
Mechanism
Crystal structures indicate that β-glucocerebrosidase binds the glucose moiety and adjacent O-glycosydic bond of glucocerebroside. The two aliphatic chains of glucocerebroside may remain associated with the lysosomal bilayer or interact with the activating protein Saposin C.[3]
Consistent with other glycoside hydrolases, the mechanism of glucocerebroside hydrolysis by β-glucocerebrosidase involves acid/base catalysis by two glutamic acid residues (E340 and E235) and precedes through a two-step mechanism. In the first step, E340 performs a nucleophilic attack at the carbon of the O-glycosidic linkage to displace the sphingosine moiety, which is simultaneously protonated by E235 as it is released from the active site. In the second step, glucose is hydrolyzed from the E340 residue to regenerate the active enzyme.[3][5]
Properties
β-Glucocerebrosidase is maximally active at pH 5.5, the pH of the lysosomal compartment.[6] Within the lysosome it remains associated with the membrane, where it binds and degrades its substrate glucocerebroside (GluCer). It requires the activating protein Saposin C as well as negatively charged lipids for maximal catalytic activity.[7][8] The role of Saposin C is not known; however, it is shown to bind both the lysosomal membrane and the lipid moieties of GluCer, and therefore may recruit GluCer to the active site of the enzyme.[9][10]
β-Glucocerebrosidase is specifically and irreversibly inhibited by the glucose analog Conduritol B epoxide. Conduritol B epoxide binds to the GCase active site, where the enzyme cleaves its epoxide ring, forming a permanent covalent bond between the enzyme and the inhibitor.[11]
Initially, GCase was thought to be one of the few lysosomal enzymes that does not follow the mannose-6-phosphate pathway for trafficking to the lysosome.[12] A study in I-cell disease fibroblasts (in which the phosphotransferase that puts Mannose 6-phosphate on proteins to target them to the lysosome is defective) showed targeting of GCase to the lysosome independent of the M6P pathway.[13] The lysosomal transporter and integral membrane protein LIMP-2 (Lysosomal Integral Membrane Protein 2) was shown to bind GCase and facilitate transport to the lysosome, demonstrating a mechanism for M6P-independent lysosomal trafficking. This conclusion was called into question when a crystal structure of GCase in complex with LIMP-2 showed a Mannose 6-phosphate moiety on LIMP-2, suggesting the complex can also follow the traditional mannose-6-phosphate pathway.[14]
Clinical significance
Mutations in the glucocerebrosidase gene cause Gaucher's disease, a lysosomal storage disease characterized by an accumulation of glucocerebrosides in macrophages that infiltrate many vital organs.[15][16]
Mutations in the glucocerebrosidase gene are also associated with Parkinson's disease.[17][18]
A related pseudogene is approximately 12 kb downstream of this gene on chromosome 1. Alternative splicing results in multiple transcript variants encoding the same protein.[19]
Drugs
Alglucerase (Ceredase) was a version of glucocerebrosidase that was harvested from human placental tissue and then modified with enzymes.[20] It was approved by the FDA in 1991[21] but has been withdrawn from the market[22][23] due to the approval of similar drugs made with recombinant DNA technology instead of being harvested from tissue. Drugs made recombinantly pose no risk of diseases being transmitted from the tissue used in harvesting, and are less expensive to manufacture.[20]
Recombinant glucocerebrosidases used as drugs include:[24]
- Imiglucerase (Cerezyme)[20]
- Velaglucerase (Vpriv)[20]
- Taliglucerase alfa (Elelyso)[25]
See also
- Closely related enzymes
References
- ↑ "Localization of ceramide and glucosylceramide in human epidermis by immunogold electron microscopy". The Journal of Investigative Dermatology 117 (5): 1126–36. November 2001. doi:10.1046/j.0022-202x.2001.01527.x. PMID 11710923.
- ↑ "Mannose 6-phosphate-independent membrane association of cathepsin D, glucocerebrosidase, and sphingolipid-activating protein in HepG2 cells". The Journal of Biological Chemistry 266 (8): 4862–8. March 1991. doi:10.1016/S0021-9258(19)67728-8. PMID 1848227.
- ↑ 3.0 3.1 3.2 3.3 "A Guided Tour of the Structural Biology of Gaucher Disease: Acid-β-Glucosidase and Saposin C". Enzyme Research 2011: 973231. 2011. doi:10.4061/2011/973231. PMID 22145077.
- ↑ "Identification and analysis of catalytic TIM barrel domains in seven further glycoside hydrolase families". FEBS Letters 544 (1–3): 103–11. June 2003. doi:10.1016/S0014-5793(03)00481-2. PMID 12782298.
- ↑ "Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate". Nature 412 (6849): 835–8. August 2001. doi:10.1038/35090602. PMID 11518970. Bibcode: 2001Natur.412..835V. http://eprints.whiterose.ac.uk/131/1/daviesgj1.pdf.
- ↑ "Secretion of human glucocerebrosidase from stable transformed insect cells using native signal sequences". Biochemistry and Cell Biology 84 (2): 148–56. April 2006. doi:10.1139/o05-165. PMID 16609695.
- ↑ "Conditions affecting the activity of glucocerebrosidase purified from spleens of control subjects and patients with type 1 Gaucher disease". Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 1041 (1): 55–63. October 1990. doi:10.1016/0167-4838(90)90122-V. PMID 2223847.
- ↑ "Identification of the binding and activating sites of the sphingolipid activator protein, saposin C, with glucocerebrosidase". Protein Science 4 (4): 756–64. April 1995. doi:10.1002/pro.5560040415. PMID 7613473.
- ↑ "Direct visualization of saposin remodelling of lipid bilayers". Journal of Molecular Biology 362 (5): 943–53. October 2006. doi:10.1016/j.jmb.2006.08.009. PMID 16949605.
- ↑ "The role of saposin C in Gaucher disease". Molecular Genetics and Metabolism 106 (3): 257–63. July 2012. doi:10.1016/j.ymgme.2012.04.024. PMID 22652185.
- ↑ "Synthesis and evaluation of glucocerebrosidase inhibitory activity of anhydro deoxyinositols from (+)-epi- and (-)-vibo-quercitols". Bioorganic & Medicinal Chemistry Letters 9 (11): 1493–8. June 1999. doi:10.1016/S0960-894X(99)00223-1. PMID 10386923.
- ↑ "LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase" (in en). Cell 131 (4): 770–83. November 2007. doi:10.1016/j.cell.2007.10.018. PMID 18022370.
- ↑ "Mannose 6-phosphate-independent targeting of lysosomal enzymes in I-cell disease B lymphoblasts". The Journal of Cell Biology 123 (1): 99–108. October 1993. doi:10.1083/jcb.123.1.99. PMID 8408210.
- ↑ "Lysosome sorting of β-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor". Nature Communications 5: 4321. July 2014. doi:10.1038/ncomms5321. PMID 25027712. Bibcode: 2014NatCo...5.4321Z.
- ↑ "Pathogenesis of Bone Alterations in Gaucher Disease: The Role of Immune System". Journal of Immunology Research 2015: 192761. 2015. doi:10.1155/2015/192761. PMID 26064996.
- ↑ "A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments". International Journal of Molecular Sciences 18 (2): 441. February 2017. doi:10.3390/ijms18020441. PMID 28218669.
- ↑ "Parkinson's disease". Lancet 386 (9996): 896–912. August 2015. doi:10.1016/S0140-6736(14)61393-3. PMID 25904081.
- ↑ PhD, Lindsey Shapiro (2023-09-18). "Researchers win Breakthrough Prize for Parkinson's genetics discoveries | Parkinson's News Today" (in en-US). https://parkinsonsnewstoday.com/news/researchers-win-breakthrough-prize-parkinsons-genetics-discoveries/.
- ↑ "Entrez Gene: GBA glucosidase, beta; acid (includes glucosylceramidase)". https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=2629.
- ↑ 20.0 20.1 20.2 20.3 "Imiglucerase in the treatment of Gaucher disease: a history and perspective". Drug Design, Development and Therapy 6: 81–106. 2012. doi:10.2147/DDDT.S14395. PMID 22563238.
- ↑ "Regulatory Matters". WHO Drug Information 5 (3): 123–4. 1991. http://whqlibdoc.who.int/druginfo/DRUG_INFO_5_3_1991_p122-125.pdf.
- ↑ "Enzyme-replacement Therapy for Lysosomal Storage Disorders". Aetna. 2014-08-08. http://www.aetna.com/cpb/medical/data/400_499/0442.html.
- ↑ "FDA Prescription and Over-the-Counter Drug Product List". U.S. Food and Drug Administration. March 2012. https://www.fda.gov/downloads/drugs/informationondrugs/ucm300963.pdf.
- ↑ "Gaucher disease and other storage disorders". Hematology. American Society of Hematology. Education Program 2012: 13–8. 2012. doi:10.1182/asheducation.v2012.1.13.3797921. PMID 23233555. http://asheducationbook.hematologylibrary.org/content/2012/1/13.long.
- ↑ Yukhananov, Anna (1 May 2012). "U.S. FDA approves Pfizer/Protalix drug for Gaucher". Chicago Tribune. Reuters. http://www.chicagotribune.com/health/sns-rt-us-fda-gaucherbre8401jz-20120501,0,5155428.story.[yes|permanent dead link|dead link}}]
Further reading
- "Mutations causing Gaucher disease". Human Mutation 3 (1): 1–11. 1994. doi:10.1002/humu.1380030102. PMID 8118460.
- "Type 2 gaucher disease: an expanding phenotype". Molecular Genetics and Metabolism 68 (2): 209–19. October 1999. doi:10.1006/mgme.1999.2918. PMID 10527671. https://zenodo.org/record/1229930.
- "Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease". Human Mutation 15 (2): 181–8. 2000. doi:10.1002/(SICI)1098-1004(200002)15:2<181::AID-HUMU7>3.0.CO;2-S. PMID 10649495. https://zenodo.org/record/1235494.
- "[Gaucher's and Fabry's diseases: biochemical and genetic aspects]". Journal de la Société de Biologie 196 (2): 135–40. 2002. doi:10.1051/jbio/2002196020135. PMID 12360742.
- "[The active site of human glucocerebrosidase: structural predictions and experimental validations]". Journal de la Société de Biologie 196 (2): 151–60. 2002. doi:10.1051/jbio/2002196020151. PMID 12360744.
- "Mutation analysis and genotype/phenotype relationships of Gaucher disease patients in Spain". Journal of Human Genetics 52 (5): 391–6. 2007. doi:10.1007/s10038-007-0135-4. PMID 17427031.
External links
- GeneReviews/NCBI/UW/NIH entry on Gaucher disease
- Glucocerebrosidase at the US National Library of Medicine Medical Subject Headings (MeSH)
- Proteopedia Acid-beta-glucosidase
PDB gallery | |
|---|---|
|







 |  |

