Pheochromocytoma is a rare tumor of the adrenal medulla composed of chromaffin cells, also known as pheochromocytes.[3] When a tumor composed of the same cells as a pheochromocytoma develops outside the adrenal gland, it is referred to as a paraganglioma.[4] These neuroendocrine tumors typically release massive amounts of catecholamines which result in the most common symptoms, including hypertension (high blood pressure), tachycardia (fast heart rate), and sweating.[5][6] Rarely, some tumors (especially paragangliomas) may secrete little to no catecholamines, making diagnosis difficult.[6] While tumors of the head and neck are parasympathetic, their sympathetic counterparts are predominantly located in the abdomen and pelvis, particularly concentrated at the organ of Zuckerkandl.[7]
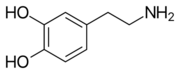
1920s: from phaeochrome (another term for chromaffin), from Greek phaios 'dusky' + khrōma 'color', + -cyte.
Signs and symptoms
The symptoms of a pheochromocytoma are related to sympathetic nervous system hyperactivity.[8] The classic triad includes headaches (likely related to elevated blood pressure, or hypertension), tachycardia/elevated heart rate, and diaphoresis (excessive sweating, particularly at night, also known as hyperhidrosis).[6] However, patients are unlikely to experience continuous symptoms. Due to the paroxysmal nature of catecholamine synthesis and release, patients may experience "attacks" or "spells" where they are suddenly overwhelmed with signs and symptoms of their tumor.[9] Attacks can occur spontaneously (without warning) or may be triggered by a variety of pharmaceutical agents (including histamine, metoclopramide, glucagon[10] and adrenocorticotropic hormone), foods that contain tyramine (cheese and wine), intraoperative tumor manipulation, intubation, or during anesthetic induction.[11]
Adrenal gland; the medulla (center, red) is the origin of the pheochromocytoma.There is an adrenal gland, highlighted in yellow, on top of each of the kidneys.
Other clinical manifestations that have been reported include (in no particular order):[5][11]
While the symptoms of a pheochromocytoma are quite common, the disease has been referred to as "the great mimic".[12] It is estimated that approximately 0.1% of patients with hypertension have a pheochromocytoma, and it is often misdiagnosed as essential hypertension.[6] As symptoms are often paroxysmal (episodic/sporadic), patients may not immediately seek treatment as the problem "disappears on its own." Furthermore, when pictured in the ideal clinical scenario (an older woman in her mid-50s), the spontaneous attacks of flushing, sweating, and a racing heart may be mistaken for pre-menopausal related hot flashes. Unmanaged pheochromocytoma is dangerous and can lead to serious complications, including death.[13][14] The cardiovascular system is the most commonly involved.[15][16][17]
In pregnancy, pheochromocytoma is associated with significant maternal and fetal mortality, mainly due to hypertensive crisis in the mother and intrauterine growth restriction in the fetus.[18][19]
Cardiovascular system
Hypertensive crisis: Pheochromocytoma-related hypertensive emergencies are one of the most feared clinical manifestations. Attacks are random and may occur secondary to a trigger (see Signs and Symptoms above) or spontaneously after a catecholamine surge.[16] The prevailing symptom is elevated systolic blood pressure (> 200 mmHg) that is unresponsive to traditional treatment regimens and threatens end-organ damage.[15] Patients require immediate, life-saving treatment to prevent further damage to other organs and/or death.
Myocardial Ischemia/Infarction: A heart attack is often caused by a significant build-up of plaque (atherosclerosis) in the coronary vessels. Patients with pheochromocytoma present with myocardial infarctions despite an overall lack of plaque build-up, indicating a different mechanism for the myocardial infarction. Current research hypothesizes that the tumor secretes massive amounts of catecholamines, which directly interact with myocardial (heart) tissue and exert negative effects including oxygen deprivation, leading to accelerated scarring and cell death.[15]
Toxic Myocarditis: Even in patients without myocardial damage, excessive catecholamines can result in abnormal ST changes on an ECG. Norepinephrine (a catecholamine) is hypothesized to result in damaged cardiac tissue by inhibiting coronary blood flow and depriving cells of oxygen, thus resulting in ischemic tissue.[17] Fortunately, following tumor excision and the subsequent quelling of catecholamines, the damage has been proven reversible.
Cardiomyopathy: Pheochromocytomas have been implicated in various types of cardiomyopathy, including (myocarditis, see above), dilated cardiomyopathy, and stress-induced or Takotsubo cardiomyopathy.[20] As with the other cardiovascular-related complications, excess catecholamines are responsible for the increased myocardial burden and significant physiologic stress.[21][non-primary source needed] Current literature indicates that most of the catecholamine-induced damage is reversible, thereby strengthening the argument for early and accurate diagnosis in order to allow for cardiac remodeling and prevent further destruction.[20][21]
Arrhythmias:Sinus tachycardia is the most common abnormal heart rhythm associated with a pheochromocytoma and is experienced by patients as the feeling of a "fluttering heart" or palpitations.[15] Many other tachyarrhythmias (fast heart rate) have also been reported.
Headache: Headaches are one of the core clinical manifestations of a pheochromocytoma and can result in debilitating pain.[6] The majority of studied patients report their pain began and ended abruptly without warning and described the pain as a severe, bilateral throbbing (although the scale of severity was not published). While 71% of the studied patients reported headaches, just over 20% of the affected patients endorsed associated nausea, vomiting, photophobia, or phonophobia, which are typically associated with migraines.[30][non-primary source needed]
Urinary system
Acute Renal Failure: Several reports have detailed rhabdomyolysis (rapid skeletal muscle breakdown) leading to acute kidney injury and the need for transient dialysis in the undiagnosed pheochromocytoma patient as their primary presenting symptom.[31][32][33][34][non-primary source needed] Kidney failure is brought about by catecholamine-induced muscle injury. Norepinephrine causes vessels to narrow, thereby limiting blood flow and inducing ischemia.[31]
Multiple organ dysfunction syndrome (MODS)[35]: Caused by an elevated inflammatory response, multiple organ dysfunction is a severe, life-threatening emergency with increasing mortality based on the number of systems involved.[36] Pheochromocytoma-related MODS is associated with multiple organ failure, hyperthermia > 40 degrees Celsius, neurologic manifestations, and cardiovascular instability resulting in either hypo or hypertension.[37] In contrast to a hypertensive crisis, pheochromocytoma-associated MODS may not respond to traditional alpha-receptor agents and may require emergent surgical excision if clinical stability is not achieved.[38]
Genetics
Current estimates predict that upwards of 40% of all pheochromocytomas are related to an inherited germline susceptibility mutation.[39] Of the remaining 60% of tumors, more than 30% are associated with a somatic mutation.[40] Given the high association with genetic inheritance, the United States Endocrine Society recommends that all patients diagnosed with a pheochromocytoma undergo an evaluation with a genetic counselor to consider genetic testing.[41] In the UK eligibility for NHS funded genetic testing is determined by criteria set by NHS England Genomics service.[42] The criteria in 2023 included all patients with paraganglioma and all patients with unilateral pheochromocytoma aged under 60.[43] The most recent data indicates that there are 25 pheochromocytoma susceptibility genes; however, just 12 are recognized as part of a well-known syndrome.[7] Determining the genetic status of a pheochromocytoma patient is crucial – each gene is inherited in a different pattern, associated with specific disease characteristics, and may respond more favorably to certain treatment options. Furthermore, early identification can guide physicians on screening recommendations for first degree relatives of patients with pheochromocytoma.[44] There is no current consensus for how and when asymptomatic carriers (individual who has a genetic variant associated with pheochromocytoma, but no current evidence of disease) should be evaluated. Conversations should occur at an individual level with the patient and their provider to develop a personalized screening plan that alternates between a biochemical (blood work) evaluation and whole-body imaging to monitor disease progression.[45][non-primary source needed]
Pediatric considerations
Additional practices may help maintain the emotional and psychological well-being of the minor. Screening includes a multidisciplinary team (endocrinologist, oncologist, psychologist, geneticist, parent, and child) where the primary focus is supporting the child.[46]
A positive result from testing during family-observed days of celebration may mask the happiness associated with these events in the future.
Testing one pediatric sibling at a time allows the family to narrow their focus when results are returned and support each sibling individually.
A negative result may be upsetting to a child if their sibling was positive; an opportunity to ask questions and process results may be helpful.
Hereditary syndromes
The following table(s) detail the clinical characteristics of the well-known hereditary pheochromocytoma gene variants[47][48][49][44][40][39][50]
Several additional gene variants have been described, but the provided information is inconsistent and a consensus has not been reached in the community if these mutations are truly pheochromocytoma susceptibility genes.[citation needed]
Diagnosis
Differential
The typical primary symptom is hypertension, which may be either episodic or continual. A diagnosis of pheochromocytoma should be suspected when the patient simultaneously presents with hypertension and the classic triad of heart palpitations, headaches, and profuse sweating.[6]
If a patient has the characteristic signs and symptoms of a pheochromocytoma and the decision is made to pursue additional biochemical (blood work) evaluation, the differential diagnosis is important as it is more likely to be something other than a pheochromocytoma given the relative frequency of 0.8 per 100,000 person-years.[2]
All patients with phaeochromocytomas are currently considered to have a lifelong risk of metastases and therefore conceptually they are all considered 'malignant'. The risk of metastasis ranges from ~5 to 15%. There is no single histological finding or biomarker to reliably predict metastatic disease, and multiparameter scoring systems have been proposed [59]
Differential Diagnosis of Pheochromocytoma by System[lower-alpha 1]
↑Misuse of over-the-counter medications such as pseudoephedrine hat are sympathomimetics
Biochemical evaluation
Gold standard
Elevated plasma free metanephrines is considered the gold standard diagnosis for pheochromocytoma.[60] Over 10 studies have confirmed that the sensitivity and specificity of this test is 97% and 93% respectively; however, there is still concern for false positive results in the correct clinical scenario.[5] When interpreting a biochemical analysis for pheochromocytoma, the provider must pay close attention to the (1) conditions of the collection, (2) all medications the patient is taking, and (3) their diet.[61]
Conditions of Collection: Unlike many routine laboratory tests that can be drawn at a moments notice, there are several recommendations that should be followed to ensure the ideal conditions and an accurate sample. Current research indicates that blood work should only be drawn after a patient has been resting supine (flat on their back) for 30 minutes before collection.[non-primary source needed][62][63] Specific supine reference values should be used in this scenario. Ensuring these conditions is difficult and may be cost-prohibitive at most institutions. In these cases, a rested, supine draw can be repeated following a positive result in a seated position to eliminate false-positive results.[61]
Pharmaceutical Interference: Many prescription, over-the-counter, and illicit substances can interfere with the proper collection of plasma metanephrines and lead to false-positive results. Providers should review a patient's medication list in-detail and have a discussion if temporarily discontinuing any of the interfering medications is possible. The most reported medications to result in falsely elevated metanephrines include: β-adrenoceptor blockers, phenoxybenzamine, tricyclic antidepressants, monoamine oxidase inhibitors, serotonin norepinephrine reuptake inhibitors (SNRI), and methyldopa.[64][non-primary source needed][61] As the majority of these medications are commonly prescribed for psychiatric conditions, a conversation with the prescriber may be necessary to facilitate alternative therapeutic options while the patient is undergoing evaluation for a pheochromocytoma.[64] After any possible prescription medications have been held, it is important to review any over-the-counter medications/supplements as well as the commonly used acetaminophen and pseudoephedrine cause false elevations in metanephrine levels.[61][64] Finally, it is important to have open, non-judgemental discussions about the patient's recreational substance use. Amphetamines, nicotine, and cocaine can result in marked plasma norepinephrine levels.
Lifestyle and Diet: As with most lab work, the patient should refrain from eating (fasting) after midnight the night prior to their collection. However, there are further recommendations specific to a metanephrines collection, including abstaining from nicotine, alcohol, and exercise for at least 12 hours prior to their lab draw.[7] Patients should also avoid catecholamine-containing foods (fruits, fruit drinks, chocolate, caffeine, tomatoes, beans, nuts, and potatoes) for a minimum of 24 hours prior to collection.[6][65][66]
While the above (3) conditions are likely to contribute to false-positive results if not controlled for, any value greater than 3 to 4 times the upper reference limit of normal should be considered diagnostic for a pheochromocytoma.[41][67]
Alternative tests
Twenty-four hour urinary metanephrines are an acceptable alternative if the plasma test is unavailable.[68] Other additional biomarkers can be helpful to aid in the diagnosis of pheochromocytoma as well, most notable is Chromogranin A. In comparison to the specificity of elevated catecholamines in the pheochromocytoma patient, chromogranin A is a non-specific polypeptide that is high in a variety of neuroendocrine tumors.[69] However, a 2006 report from Italy found that over 90% of studied pheochromocytoma patients demonstrated elevated chromogranin A levels.[70] If metanephrine values are equivocal, chromogranin A can be used as an adjunct marker to predict the presence of a tumor.[citation needed]
Borderline elevated metanephrines present a diagnostic challenge to the physician – the first step is to repeat the labs, taking extra precautions to follow the gold standard diagnosis described above, including the conditions of collection, pharmaceutical interference, and any potential diet and lifestyle habits that could alter the results. If the offending medications cannot be discontinued or repeated labs remained the same, consider administering a clonidine suppression test.[6][71] In the 1970s, the drug clonidine hydrocloride swept the market as a novel agent for hypertension; however, the reported side-effects (nausea, vomiting, drowsiness, dryness of the eyes and mouth, constipation, and generalized weakness) limit compliance and have vastly diminished prescriptions.[72] While the adverse side-effects with clonidine are inconvenient, the most dangerous aspect of clonidine is withdrawal rebound hypertension – that is, when the medicine is abruptly discontinued, blood pressure may rapidly return or surpass the original value.[73][74][75] However, a one-time, weight-based dose can be utilized in limited settings to help determine disease status.[61] After fasting overnight, patient's will present to their testing site for a baseline metanephrines blood draw and clonidine administration. They will remain supine for (3) hours and a repeat blood draw will be taken. A positive result (indicating a pheochromocytoma) will occur if the plasma metanephrine levels remain elevated after clonidine is given. If the results are the same or fall, the test is negative and the patient does not have a pheochromocytoma.[61] It is important to note that if a patient does not have a pheochromocytoma, they may become extremely hypotensive following clonidine. Patients should not depend on themselves for transport following this test.
Plasma methoxytyramine is a breakdown product of the catecholamine, dopamine. Paragangliomas of the head and neck commonly secrete dopamine, but are referred to as "biochemically silent" because they do not cause the characteristic symptoms associated with a pheochromocytoma. However, methoxytyramine can be utilized to detect the tumors of the head and neck.[non-primary source needed][76] Further research indicates that the biomarker is also a useful indicator of metastatic disease – which is the only current biochemical evidence of metastases to date.[77]
Biochemical phenotypes
Structure of epinephrine
While diagnostic, laboratory values can also provide physician's with important information about the type, location, size, and associated tumor genotype.[67] There are (3) major, well-recognized biochemical phenotypes that can be used by health care providers to direct patient care.[78]
When plasma metanephrine levels were elevated to greater than 15% of the combined levels of normetanephrine and metanephrine, an adrenal tumor or a recurrence of an adrenal tumor that had already been excised can be predicted
Patients are more likely to present with the classic, paroxysmal (episodic) symptoms described above[67]
Structure of norepinephrineNoradrengeric (Norepinephrine and normetanephrine)
More likely to indicate an extra-adrenal tumor[79]
Patients are more likely to present with continuous, persistent pheochromocytoma-related symptoms (hypertension and tachycardia) compared to those that are classically episode with an adrenergic phenotype[67]
Structure of dopamineDopaminergic (Dopamine and 3-methoxytyramine)
More likely to indicate an extra-adrenal tumor of the head and neck[78]
Patients are more likely to be asymptomatic; however, they may present with non-specific signs of nausea, vomiting, abdominal pain, diarrhea, and weight loss secondary to the stimulation of dopamine receptors throughout the gastrointestinal tract[67]
Across both an adrenergic and a noradrenergic phenotype, the greater the sum of plasma or urinary concentrations of metanephrine and normetanephrine, the larger the expected tumor diameter.[79]
Tumor localization
Anatomic imaging
Anatomic imaging refers to computed tomography (CT) [CAT scan] or magnetic resonance imaging (MR) scans. These imaging modalities serve to initially locate the tumor and provide detailed information about size, morphology, and structural relation to adjacent internal structures.[80] Traditionally, a patient presents to their physician for symptoms concerning for a pheochromocytoma, which prompts a biochemical evaluation. If the results are positive, the patient is referred for anatomic imaging with a CT or MR scan. However, as anatomic imaging becomes more readily available, patients are referred to an endocrinologist after an incidental (unanticipated finding) adrenal nodule is found on a scan ordered for another reason.[81] For example, "Patient M" presents to his local emergency room for abdominal pain and a CT is ordered to rule-out appendicitis; however, the radiologist notes there is a 3.5 centimeter right adrenal mass.[citation needed]
While there has not been a consensus on if CT or MR is the preferred imaging modality in pheochromocytoma, each method has its associated strengths and weaknesses. As CT expose the patient to ionizing radiation, MR is preferred in children and pregnant women.[82] Furthermore, the intravenous contrast used in CT can cause kidney damage and should therefore be avoided in patients with pre-existing damage.[83] However, patients who struggle with being in confined spaces for extended periods of time (claustrophobia) cannot often tolerate an MR as the machine is close-ended compared to the open-ended design of a CT.[84] When patients become anxious and begin to move in the machine, this causes motion artifact, which occurs less in CT-based images.[85]
Compared to CT and MR, ultrasound is not a preferred imaging modality and should be avoided in the pheochromocytoma patient. However, in specific patient populations where avoid ionizing radiation is the top priority (children, pregnant women), ultrasound can be used as an adjunct method when MR may be unavailable or the patient is unable to complete the scan. Furthermore, if an acute adrenal hemorrhage is suspected in a pheochromocytoma patient, ultrasound is a quick, painless, radiation-less, and cheap modality for a "first-pass" before the above imaging modalities or surgery is used to confirm the diagnosis.[86]
Functional imaging
The imaging modalities discussed below are for tumor characterization, confirmation of metastatic disease, and treatment planning – they are not used to discern tumor location or help the surgical team prepare for excision.[87] For most pheochromocytoma patients, functional imaging will follow a CT or MR. If anatomic imaging only demonstrates an adrenal tumor without evidence of disease anywhere else in the body and the metanephrine levels are overtly elevated, functional imaging can be foregone in favor of prompt surgical excision.[82] Over the last decade, there have been five functional techniques used to evaluate the pheochromocytoma patient (1) 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET), commonly referred to as the PET scan, (2) iodine-123 meta-iodobenzylguanadine (123I-MIBG), (3) 18F-flurodihydroxyphenylalanine (18F-FDOPA),(4) 68Ga-DOTA coupled somatostatin analogs (68Ga-DOTA),(5) 11C-Hydroxy ephedrine(HED-PET). From this point forward, these imaging modalities will be referred to in their abbreviated names found in parentheses.[citation needed]
MIBG Scintigraphy – the pheochromocytoma is appreciated in the left panel on the right side of the screen (right panel; left side of the screen) as the darkened circle towards the abdomen. The darkened structure at the head of the patient is the thyroid gland, while the darkened structure in the pelvis of the patient is the bladder. This is normal physiologic uptake.
The first functional imaging technique utilized in pheochromocytoma patients was 123I-MIBG scintigraphy. Given the compounds similar structure to the catecholamine norepinephrine (secreted by pheochromocytomas), MIBG was well-suited for uptake by most neuroendocrine tumors.[88] Furthermore, if a patient was found to be positive on an MIBG scan, they were eligible for MIBG treatment, offering additional avenues for those with widespread metastatic disease.[89] However, further investigation revealed that while MIBG excelled with adrenal lesions, it was far less superior in patients with extra-adrenal paragangliomas, particularly with specific genetic variants like succinate dehydrogenase subunit X (SDHx).[77] As the positron emission tomography scans were developed, MIBG has slowly loss its favor for the pheochromocytoma patient.[77]
Error creating thumbnail: Unable to save thumbnail to destinationFDG PET – the tumor is appreciated as the dark structure in the patient's left chest. Darkened structures at head of patient is brain, in the abdomen are the kidneys, in the pelvis is the bladder. These are normal.
Of the four above mentioned modalities, 18F-FDG PET is the most common and readily available functional imaging technique at most hospital systems, but the least-specific to neuroendocrine tumors (Image Left). In 2012, over 200 patients participated in a trial that compared the current gold standard of the time (MIBG/CT/MRI) to the novel FDG PET. Compared to its functional counterpart, FDG outperformed MIBG in detecting soft-tissue and bone metastases with higher specificity in patients with biochemically active tumors.[77]
Following the development of FDG-PET, neuroendocrine-specific PET scans began to emerge. One of the first favorable imaging modalities was 18F-FDOPA, which demonstrated a high sensitivity in detecting head and neck paragangliomas as well as non-metastatic disease outside of the head and neck.[77][90] Unfortunately, in cases of metastatic disease, particularly related to succinate dehydrogenase subunit B (SDHB) mutations, 18F-FDOPA fell inferior to the traditional FDG-PET.[91] However, for patients with genetic variants in other pheochromocytoma-susceptibility genes (NF1, VHL, RET)18F-FDOPA has become the preferred radiopharmaceutical agent.[92]
The newest PET modality involves somatostatin receptor type two receptor imaging with 68Ga-DOTA analogues.[85] Over the last decade, further research continues to indicate the superiority of this functional imaging modality in a wide range of clinical scenarios, even surpassing anatomic imaging (CT/MR) in pediatric patients with succinate dehydrogenase (SDHx) mutations.[non-primary source needed][93] While FDOPA inconsistently detected metastatic disease, 68Ga-DOTA analogues have demonstrated superior localization of metastatic pheochromocytoma.[non-primary source needed][94] When directly compared in one head-to-head study in 2019, 68Ga-DOTA analogues outperformed FDOPA, particularly in the detection of metastatic bone lesions.[95] An additional benefit of the DOTA analogues is the ability for treatment with peptide receptor radionuclide therapy, which will be discussed in the treatment section below.[96]
Also, HED-PET has shown to be an accurate tool to diagnose and rule out pheochromocytoma in complex clinical scenarios and to characterise equivocal adrenal tumours.[97]
Management
Surgery
Surgical resection is the only curative option for pheochromocytoma as of 2019.[98] A successful excision is a multidisciplinary effort involving the endocrinologist and the patient pre-operatively (discussed below) and the surgical team and anesthesiologist intraoperatively. Without frequent and adequate communication between all of the above-mentioned teams, a favorable outcome is much more difficult.[98] The United States Endocrine Society 2014 Clinical Practice Guideline for pheochromocytoma recommend a laparoscopicadrenalectomy (minimally invasive technique) for most adrenal tumors, unless they are invasive or are larger than 6.0 centimeters.[41] A 2018 systematic review suggests that laparoscopic retroperotenial adrenalectomy appears to reduce late morbidity, time to oral fluid or food intake and time to ambulation when compared to laparoscopic transperitoneal adrenalectomy, however there is uncertainty about these effects due to very low-quality evidence.[99] For outcomes such as all-cause mortality, early morbidity, socioeconomic effects, and operative and postoperative parameter, the evidence is uncertain about the effects of either interventions over the other.[99]
It is important to note that larger tumors even for those larger than 6.0 cm can be attempted with a minimally invasive approach, but the team should be prepared to convert to an open procedure if necessary.[non-primary source needed][100][101] An open procedure (traditional surgical technique) is currently preferred for extra-adrenal disease, unless the tumor is small, non-invasive, and in an easy to maneuver location. While previous data indicated the need for a minimally invasive approach with malignant and/or metastatic disease, current research indicates a successful operation is feasible and results in a shorter hospital stay.[non-primary source needed][102] Literature within the last decade has also demonstrated that the robotic technique may be successfully utilized for adrenal tumors.[103]
Typically, complete or total adrenalectomy is performed; however, a technique referred to as "cortical-sparing" can leave a remnant (piece) of the adrenal gland in hopes of avoiding life-long steroid replacement if the left and right adrenal glands need to be removed.[104] The issue is particularly important in patients with MEN and VHL-related disease, which has a higher chance of bilateral pheochromocytomas.[non-primary source needed][105] The risk of leaving adrenal tissue is recurrent disease (tumor comes back). A 2019 cohort study reported that despite a 13% recurrent rate in patients who underwent a cortical-sparing adrenalectomy for pheochromocytoma, there was no decreased survival compared to their total adrenalectomy counterparts.[104]
Pre-operative management
Arguably, the most important part of a pheochromocytoma surgical plan is an adequate pre-operative blockade. Excess catecholamines have been described as a dormant volcano, ready to erupt at any time, wreaking catastrophic havoc on the body.[106] While an eruption can occur at any time, two of the most common triggers are anesthesia and direct tumor manipulation, making surgery one of the most dangerous times for a pheochromocytoma patient if not properly prepared.[non-primary source needed][107] In order to help circumvent a catecholamine-crisis, the United States Endocrine Society recommends that all patients with functional (hormonally active) tumors be started on a pre-operative alpha-adrenoceptor blockade a minimum of seven days prior to surgery.[41] There are several medication options depending on the clinical scenario, each with their own associated strengths and weaknesses.
Alpha blockade
If the patient's blood pressure is moderately elevated, a selective, short-acting alpha-1 adrenoceptor antagonist (doxazosin, prazosin, terazosin) is the preferred agent.[106] However, the patient should be warned about the potential side-effect known as "the first-dose phenomenon." When patients are initially exposed to one of the above agents, they may become lightheaded, dizzy, and nauseous, particularly when transferring from a seated to standing position due to a rapid decrease in blood pressure.[108] These effects will decrease with time, but providers can try to avoid them by starting at a low-dose and slowly increasing until they reach their desired amount. In patient's with uncontrolled hypertension, the non-selective alpha-1 and 2 adrenoceptor antagonist (phenoxybenzamine) should be utilized.[106] Unfortunately, compared to the selective agents listed above, phenoxybenzamine is much more expensive and may not be readily available to some patients. Common side effects include dry mouth, nasal congestion, and impaired male ejaculation, all of which do not cease with time and may limit patient compliance.[109] While uncommon, patients may have a hormonally-active pheochromocytoma and a normal blood pressure. One comparison from 2014 found that a small dose of a calcium-channel blocker (such as amlodipine) may be used pre-operatively in some people.[110] This will not drastically lower the patients blood pressure and make them hypotensive, but it will assist the surgical and anesthesia teams if there is hemodynamic instability during the operation.
Beta blockade
An elevated heart rate (tachycardia) and the feeling of a racing heart (palpitations) may follow after initiating an alpha-adrenoceptor antagonist. If that is the case, a beta-adrenoceptor antagonist is then prescribed to control the heart rate.[106] Just as with the alpha antagonists, there are selective (beta-1) and non-selective (beta-1 and beta-2) adrenoceptor antagonists. The selective agents (atenolol, metoprolol) are preferred to the non-selective agents (propranolol).[106] There are several (labetalol, carvedilol) combined alpha-beta-adrenoceptor antagonists. These agents should be avoided whenever possible as there is upwards of seven times more beta-adrenoceptor antagonism than alpha, which can worsen hypertension and lead to a catecholamine crisis.Template:Outdated inline[111]
Complications
Beta-adrenoceptor antagonists should not be given alone in a pheochromocytoma patient – this can lead to severe consequences.[non-primary source needed][112] In 1995, a team of physicians from London described the death of a person who had been recently diagnosed pheochromocytoma after initiation of propranolol, a non-selective beta blocker. She quickly developed a hypertensive crisis leading to shock, myocardial infarction, heart failure, and dense right hemiplegia. Despite attempts at resuscitation, the person died several days later.[113] This complication is related to the impact that alpha and beta-adrenoceptor antagonists have on blood vessels combined with the actions of catecholamines. The normal blood vessel is open, allowing for adequate blood flow. When catecholamines activate the alpha receptor, the vessel constricts (gets smaller), which results in hypertension.[114] However, when catecholamines active the beta receptor, the blood vessel dilates (gets larger) and allows for increased blood flow, reducing the blood pressure.[115] If a pheochromocytoma patient is only started on a beta-adrenoceptor antagonist, this reverses the protective vasodilation and worsens the patient's hypertension.
Controversy
While the pre-operative alpha and beta blockade discussed above is overwhelmingly recognized as the standard of care, particularly in the United States, there has been discussion at the international level if alpha-blockade is necessary. In 2017, a team of researchers from Germany published an observational case series that called into question the current recommendations for alpha-blockade.[116] The study examined the intraoperative maximal systolic arterial pressure in people with and without alpha-adrenoceptor blockade and found no difference in complications between the two groups.[116] The following year, a group from France published a similar article with a warning against waiting an entire week to begin alpha-blockade. The French researchers called for immediate surgical intervention and consideration of steps to mitigate any intraoperative catecholamine crisis.[117] These articles resulted in rebuttals[107][118] from research teams in the United States, but an international consensus has not yet been reached.
Perioperative fluid status
Excess catecholamines cause a decrease in the total blood volume, making a patient vulnerable to hypotension during the operation.[119] Therefore, a high-sodium diet with adequate fluid intake should be encouraged prior to surgery.[120] Some institutions in the United States will even admit patients the night prior to surgery for intravenousfluid replacement starting at midnight until the time of the operation.[106] However, a small trial from 2009 reported no difference in mortality in patients treated with preoperative intravenous fluids compared to those who did not.[121]
In a 2010 survey of 40 endocrinologists by researchers at the Cedars-Sinai Medical Center in Los Angeles, California, nearly all indicated the importance of preoperative volume resuscitation (having the patient take in plenty of fluids prior to surgery). However, after reviewing their patient data, over 60% of the same physicians failed to discuss salt-loading and adequate hydration.[needs update][non-primary source needed][122] When the patients were stratified by age, those that were younger received the advice to hydrate, but older patients did not. It was hypothesized that the providers chose to forego volume repletion in the older patient population for fear of their potential comorbidities (heart failure) where excess fluid is dangerous.[122] While there is still no recognized consensus or gold standard, providers should individualize the decision based on the patient's perceived nutritional standing, volume status, comorbidities, and ability to self-hydrate.
Post-operative management
Histopathology on the resected tumor confirms the diagnosis, by typical features as shown
The most common post-operative complications, likely causes, and treatment options are:[123][124]
Hypertension: In the pheochromocytoma patient, postoperative hypertension could indicate incomplete tumor resection or another tumor of unknown location. However, the traditional, non-specific causes of postoperative hypertension including pain, fluid overload, and essential hypertension must also be considered. A perioperative hypertensive crisis is first treated with a 5.0 milligram (mg) intravenousbolus of phentolamine, with additional 5.0 mg dose every ten minutes until the blood pressure falls within an acceptable range.[non-primary source needed][125] If the blood pressure is only minimally elevated, the patient can resume their alpha and beta-adrenoceptor antagonist from prior to surgery.[123]
Hypotension: There are several reasons a patient may have low blood pressure in the post-operative period. First and foremost, the tumor (and its abundance of catecholamines causing high blood pressure) has been removed. Furthermore, the patient may still experience the effects of their alpha-adrenoceptor antagonist, which causes lower blood pressure.[124] First-line treatment for postoperative hypotension is aggressive fluid resuscitation, which is why ensuring the patient is well-hydrated (see above) prior to surgery is so imperative.[123]Vasopressors may be needed if the blood pressure does not respond to fluids.
Endocrine
Hyperglycemia: Catecholamines prevent the secretion of insulin – a hormone responsible for lowering the body's blood glucose (sugar). Blood glucose levels should be checked frequently in the perioperative period and insulin should be given as needed if levels are elevated. Following resection, tumor-related hyperglycemia is likely to resolve.
Hypoglycemia: After the tumor is removed, insulin is no longer inhibited, which can bring the blood glucose dangerously low. Symptoms include tremor, anxiety, palpitations, sweating, altered mental status (confusion), dizziness, and blurred vision.[126] A retrospective analysis of beta blocker found that some beta blocker use may cause people to more prone to hypoglycemia and not experience these symptoms, which could delay the diagnosis.[127]
Adrenal Insufficiency: Following a bilateral adrenalectomy (left and right), the patient is no longer capable of secreting the necessary hormones to keep their body functioning. Life-long steroid (hydrocortisone and fludrocortisone) oral supplementation may be required to ensure they do not develop adrenal insufficiency.[non-primary source needed][128] When the body is stressed (during surgery), the adrenal glands naturally produce more steroids; however, if the glands have been removed, they are unable to do so. Therefore, "stress-dosing" steroids are required and should be started intraopertively to mimic the natural physiology of the adrenal glands.[129] The typical regimen when post-operative adrenal insufficiency is thought to be likely:[123][124]
Repeat administration of 25–50 mg intravenous hydrocortisone every eight hours for a maximum of 72 hours (3 days) after the operation. Convert to oral replacement therapy as soon as the patient is able to take medication by mouth
Patients should be transitioned to a normal maintenance (regular, daily) dose of steroids prior to discharge and referred to endocrinology for proper titration and management. Depending on the patient's total body surface area, the total typical daily dose of hydrocortisone is between 15 and 25 mg daily (divided into morning and afternoon pills).[130]
Those who have lost both their adrenal glands will also require another steroid (mineralcorticoid replacement). The typical daily dose is between 50 and 200 micrograms of fludrocortisone[130]
There have been many other reported complications (renal failure, heart failure, intestinal pseudo-obstruction) following tumor resection. However, the above are more likely to be encountered, which is why their management has been specifically outlined here in this article.
Metastatic disease
Diagnosis and location
Metastatic pheochromocytoma is defined as the presence of tumor cells (chromaffin tissue) where they are not normally found.[131] Patients with a paraganglioma are more likely to develop metastases than those with a pheochromocytoma.[132] The most common extra-adrenal sites of metastases are the lymph nodes, lung, liver, and bone.[133] There have been several studied risk factors associated with the development of metastatic disease – while the patients genetic background plays an important role, the initial age of presentation and size of the tumor lead to negative outcomes.[131] Of all the genetic variants, succinate dehydrogenase subunit B(SDHB) mutations have the highest rates of developing metastatic disease.[132] Another study has reported increased mortality associated with male sex and synchronous metastases.[132] Metastases are divided into synchronous and metachronous; those that are synchronous have developed within several months of the primary tumor, while metachronous metastases do not appear for a significant period of time.[134]
Laparoscopic approach to the original disease, especially in big tumors, has been appointed as an important risk factor for tumoral seeding.[135]
Despite all of the below potential treatment options, recent literature highlights that (for most patients) metastatic pheochromocytoma is slow-growing. In patients with minimal disease burden, a "watch and wait" approach with frequent imaging to monitor disease is favorable, withholding treatment until evidence of progression is visualized.[136]
Surgery – Normally, the goal of surgery is complete cytoreductive surgery;[135] leave no remnant of disease.[137] However, with widespread metastatic disease, this is not always feasible. Therefore, a surgical debulking procedure is performed (removing as much of the cancerous tissue as possible) in order to reduce patient symptoms by removing the source of catecholamines, improve response to chemo or radionuclide therapy, or simply decrease the size of the tumor.[138] Unfortunately, the intended relief from the procedure is often short-lived, especially if the patient has disease outside the abdomen.[138] A 2013 study from the National Institutes of Health reported that a majority of patients with recurrent biochemical evidence of disease within one year of the operation and less than 30% continued to be biochemically free of disease after five years.[138]
In contrast to an operation for non-metastatic disease, an open procedure may be preferred over a minimally invasive technique in order to circumvent potential tumor spread.[139] This also aids surgical visualization and offers the best opportunity to identify and remove metastatic lymph nodes.[140] Reports have also indicated the utility of administering a radionuclide agent like iodine-123 meta-iodobenzylguanadine (123I-MIBG) prior to surgery and then scanning the patient intraoperatively with a probe to detect disease that may be missed with the naked eye.[141]
Patient receiving radiation therapy to the region of the head and neck. Full facial mold is in place to protect areas where they do not want exposure.
Radiation Therapy – With regard to pheochromocytoma, radiation techniques are primarily used for pain control, specifically with regards to bone metastases, local control of the disease, and to limit spinal cord compression.[142] A multidisciplinary team from the Mayo Clinic retrospectively reviewed all of their patients who underwent external beam radiation therapy from 1973 to 2015 and reported that 94% of patients acknowledged symptomatic improvement and over 80% of patients showed no evidence of recurrent disease five years post-therapy.[143] Another report from the same institution looked at almost two decades of patients who underwent radiofrequency ablation, cryoablation, or percutaneous ethanol injection for metastatic pheochromocytoma and reported that local control was achieved in over 85% of targeted lesions and that 92% of procedures were associated with reduced pain and/or symptoms of catecholamine excess.[144]
Chemotherapy – The most common chemotherapy regimen for metastatic pheochromocytoma is cyclophosphamide, vincristine, and dacarbazine, collectively known as CVD.[145][146] Response to therapy is measured by a reduction in total tumor volume as well as symptomatic relief, reported by the patient. A systematic review and meta-analysis of unstratified pheochromocytoma patients who underwent CVD therapy showed that 37% of patients had a significant reduction in tumor volume, while 40% of patients experienced lower catecholamine burden.[145] While there was no difference in overall survival between patients whose tumors shrunk versus those without a response (no reduction in tumor burden via imaging), even in non-responders, patients reported feeling better, blood pressure was lower, and some patients were even able to undergo surgery following disease stabilization with CVD.[147] When patients are studied by various categories, research has suggested that females are less likely to have extended survival with CVD chemotherapy compared to their male counterparts.[148] Genetic status has been shown to greatly impact response to CVD. A team of researchers from the National Institutes of Health reported that patient's with succinate dehydrogenase subunit B(SDHB) mutations are not only more likely to initially respond to CVD, but that they also experienced over 30 months of progression-free survival (time until tumor returned) with continued administration.[149]
However, CVD is not the only proven chemotherapeutic regimen in the pheochromocytoma patient. A 2018 report demonstrated the remarkable response of two SDHB patients who failed CVD chemotherapy (disease progressed despite medication), but were then treated with temozolomide (TMZ) and had progression free survival of 13 and 27 months, indicating that TMZ can be considered as an alternative treatment regimen in those who have progressed on CVD.[150] Several studies have since reported successful responses with TMZ, particularly in the SDHB sub-population.[151][152]
Radionuclide Therapy
Iodine-131 meta-iodobenzylguanadine (MIBG)
As was mentioned in the functional imaging section above, MIBG is not only useful in locating the presence of metastatic disease, but also as an available treatment modality. In 2019, a multi-center phase 2 trial looked at the safety and efficacy of MIBG therapy in metastatic or unresectable (not conducive to surgery) pheochromocytoma patients and the results were promising.[153]Median overall survival was 36.7 months and 92% of patients had at least a partial positive response (tumor shrinkage) or stable disease without progression within the first year of the study. Furthermore, over a fourth of the patients were able to decrease their anti-hypertensive medications and reported symptomatic improvement.[153] There are several patients who are not eligible for MIBG treatment, including pregnant women (exposure to radiation is harmful to the fetus), women who are actively breast feeding, patients in renal failure, and those are who not expected to live longer than three months.[154] As MIBG therapy can destroy the thyroid, protective medications (potassium iodide) are started prior to treatment and need to be continued for at least three weeks after therapy concludes.[154] Associated side effects (muscle weakness, nausea, vomiting and hematologic (blood) toxicities, are common, but often minimal, and can be mitigated with slow, steady dosing.[155]
Top: Purple lesions are metastatic disease detected with DOTATATE imaging. Bottom: Same patient. Purple lesions are metastatic disease detected with FDG PET
The newest of the treatment options, PRRT utilizes the 68-Ga DOTA analogues mentioned above in the functional imaging section.[156] Treatment with 177Lu-DOTATATE first demonstrated success in patients with undifferentiated neuroendocrine tumors and then trials began with metastatic pheochromocytoma patients.[157][158] In 2019, Vyakaranam et al. published favourable results for their 22 patients who underwent PRRT, with partial response in 2 patients and stable disease (no progression) in the remaining 20 patients.[159] Overall toxicity was low, with no high-grade haematological (blood) or kidney damage reported.[159] At the end of that same year, a systemic review looked at all published articles (12) where metastatic pheochromocytoma patients underwent PRRT and found that treatment-related adverse events are minimal, with only 5 out of 102 patients choosing to voluntarily initiate treatment discontinuation.[160] Newer reports have detailed the utility of combining 90Y-DOTATATE with the traditionally studied 177Lu analog and the various possibilities and novel treatment options these combinations will bring to the field.[96] While the overall reported side-effects have been promising, it is important to note that a collaborative effort between the National Institutes of Health and Radboud University Medical Centre reported two unfortunate cases of rapid disease progression following a remarkable, almost complete response to PRRT. While the etiology of their recurrence is unknown, the team speculated that an elevated tumor marker (Ki-67) could be an indication of a poor response to PRRT and called for pre-PRRT assessments to include Ki-67 values to help individualize patient treatment plans.[161]
Prognosis
According to the National Cancer Institute, prognosis is defined as the likely outcome of a disease OR, the chance of recovery or a recurrence.[162] This is an extremely difficult question when it comes to pheochromocytoma, and the answer depends on the patients genetic status, presence of metastatic disease, and the location of their primary tumor.[163] An article about prognosis published in 2000 reported a 91% 5-year survival rate in their patient population; however, over 86% of their patients had sporadic tumors (no known genetic mutation), which commonly have low malignant potential.[164] In 2019, a consortium of almost twenty European medical centers looked at the prognosis of malignant pheochromocytoma and the data starkly varies from the report of sporadic, single tumors, with a median survival of 6.7 years.[165] Overall survival improved if the patient had (1) disease of the head and neck compared to abdomen, (2) less than 40 years of age, (3) and if their biochemistry was less than five times the upper reference limit of normal.[165]
Recent literature has detailed several factors that predict accelerated progression of disease and higher mortality rates, including patients who choose to forego surgical resection of their primary tumor, larger tumors at initial presentation, older age at initial diagnosis, and a shortened time from primary tumor to presence of metastases.[166] The actual location of the metastases can also indicate prognosis, with osseous lesions (bone) faring better than their soft-tissue (lung, liver) counterparts.[167]
Epidemiology
According to the North American Neuroendocrine Tumor Society, the prevalence of pheochromocytoma is between 1:2,500 and 1:6,500, meaning that for every 2,500–6,500 people, there is (on average) one person with pheochromocytoma.[168] In the United States, this equates to an annual incidence (new cases per year) of 500 to 1,600 cases.[168] However, approximations in the early 2000s reported that upwards of 50% of pheochromocytoma diagnoses are at autopsy; therefore, the above estimations may be lower than expected.[11] In a 50-year autopsy case series, the Mayo Clinic reviewed 54 pheochromocytoma cases between 1928–1977 and discovered that just 24% of the patients were correctly diagnosed prior to their death.[needs update][non-primary source needed][169] Outside of the United States, several countries have documented their own epidemiological studies and compared them to what is known in North America. In the first national, epidemiological population-based study in Asia utilizing Korean National Health Insurance Service data, the prevalence of a pheochromocytoma was reported at 2.13 per 100,000 persons with an incidence of 0.18 per 100,000 person-years.[170] This is lower than the occurrence reported from Rochester, Minnesota (0.8 per 100,000 person-years), in a study conducted from 1950 to 1979.[2] However, the Netherlands also conducted a study using a nationwide registry and reported incidence results of 0.57 per 100,000 person-years from 2011 to 2015, which was a significant increase from their 0.37 cases per 100,000 person-years reported from 1995 to 1999.[171] Current hypotheses for why the incidence of pheochromocytoma is growing in the Dutch population point to the advent of modern imaging evaluation and the ability to detect these tumors prior to death.[172] While each of the above studies reported varying incidence and prevalence values, all have indicated that the average age at initial diagnosis is between the third to fifth decade of life.[173] When younger patients are diagnosed with a pheochromocytoma, there should be a high suspicion for hereditary disease, as genetic anticipation (earlier disease onset with each generation) is associated with some mutations.[174]
Likelihood of diagnosis when an adrenal-nodule is identified; pheochromocytoma is in yellow near the top-right corner.
Classically, the pheochromocytoma "rules of 10" have been taught, particularly to medical students:[175]
10% of patients have malignant disease
10% of patients have bilateral (both left and right adrenal glands) disease
10% of patients have extra-adrenal (paraganglioma) disease
10% of patients have inherited (familial disease)
Despite the prominence in many respected textbooks, these guidelines have since been established as inaccurate and are not used in current epidemiological discussions.[173]
As suggested above, incidental imaging has become a major player in the diagnosis of patients with pheochromocytoma, with current estimates that 10–49% of all cases diagnosed after imaging was obtained for another reason. When an adrenal nodule (potential tumor) is discovered on computed tomography or magnetic resonance imaging, there is a 5–10% chance the lesion is a pheochromocytoma.[173] The incidence of adrenal tumors is found in the infographic above, with pheochromocytoma noted in yellow in the top right corner.
History
Professor Ludwig Pick, the German physician who first coined the term "pheochromocytoma" in 1912 after recognizing the color-change associated with the addition of chromium salts
In 1800, an Irish physician (Charles Sugrue) penned a case report to the London Medical and Physical Journal describing the peculiar case of an 8-year-old male patient who had had seemingly random fits of pain concentrated in the abdomen accompanied by "a hectic flush distinctly marked on each cheek" with a "constant profuse and universal perspiration."[176] Following his death, a group of physicians performed an autopsy to determine cause of death and discovered a six-inch oblong tumor composed of an unknown "yellow-ish coloured substance" coming from the capsula renalis (what is now known as the adrenal gland).[176] This would become the first known clinical description of a pheochromocytoma, but as no features of the tumor itself were described, complete credit is given to the Germany Felix Fraenkel, who provided a clinical and morphologic picture of this tumor.[177][178] While various physicians were recognizing symptoms and treating patients, Czech biologist Alfred Kohn reported his discovery of the paraganglia system, which would later become crucial to the diagnosis of these tumors. Furthermore, he also introduced the term "chromaffin," allowing pathologists to recognize tumors that arose from the adrenal gland.[179]
In 1908, two pathologists, Henri Alezais and Felix Peyron, introduced the scientific community to "paraganglioma" after they discovered extra-adrenal tissue that reacted to chromium salts, which mimicked the reaction of the adrenal medulla.[180] Just four years later, German pathologist Ludwig Pick coined the term "pheochromocytoma" after he observed the consistent color change in tumors associated with the adrenal medulla.[181] Many surgeons attempted to remove these tumors over the next decade, but their patients died intraoperatively from shock. In 1926, Charles Mayo (a founder of the Mayo Clinic) became the first physician to successfully excise a pheochromocytoma.[181] However, Mayo was likely unaware of the diagnosis prior to the operation. Not until 1929 was a pheochromocytoma recognized preoperatively.[11] Throughout the early 1900s, the operative mortality rate for a pheochromocytoma ranged from 30 to 45%. Retrospective series have postulated that these alarmingly high death rates were due to the lack of a pre-operative blockade with alpha and beta-adrenoceptor antagonist and the need for modern anesthesia practices.[182] From this point forward, physician-scientists have been recognizing patterns in patients with pheochromocytoma and identifying genetic associations and various syndromes.[11]
Society and culture
While a rare disease, there have been several references to pheochromocytoma in popular culture and the media, specifically medical television dramas. Additionally, there is a strong online patient advocacy community that works to connect patients with rare diseases and allows them to meet other individuals who are experiencing similar diagnoses and treatment strategies.
Zebra culture
The zebra has become a powerful symbol in the pheochromocytoma advocacy community and represents the rare medical cases that are more likely to be misdiagnosed.
In the medical community, students are often taught "when you hear hoofbeats, think horses, not zebras."[183] In other words, common diagnoses are common, so healthcare professionals should first rule out what is most expected (the horses) before diving into the rare etiologies that are far less likely to be correct (the zebras). However, the symbol of the zebra has become increasingly powerful to the rare disease community and resulted in several organizations, societies, and special events (Rare Disease Day) to draw attention to the least common option sometimes being the correct diagnosis.[184]
The National Organization for Rare Disorders is a United States–based advocacy parent organization with the goal of promoting awareness and research opportunities to cure rare diseases.[185] Groups such as these encourage patients to become their own advocates and change agents in their healthcare decision-making processes.
Media
In July 2012, an actual pheochromocytoma patient, Tannis Brown, former vice-president of the PheoPara Troopers, was featured on the Discovery Fit & Health Network program Diagnosis: Dead or Alive.[186] The show highlighted her personal struggle with misdiagnosed disease as many physicians felt her episodic headaches and hypertension (high blood pressure) were related to stress.[187]
In the seventh and eighth seasons of Grey's Anatomy, series regular Henry has a Von Hippel-Lindau(VHL) mutation that has resulted in a pheochromocytoma. The story arc was met with mixed opinions from the rare disease community.[188] The executive director of the VHL Alliance was happy with the portrayal of a VHL patient in mainstream media, but pointed out that of the four scripts she knew of with a VHL patient, three involved a pheochromocytoma, which occurs in less than a fifth of all VHL patients.[189][190]
A case of pheochromocytoma was featured in the first episode of season 2 of House MD. Dr. House and his team are tasked with diagnosing and treating an inmate on death row. Although the patient has a violent history of homicide, Dr. House suspects that his episodic rage and aggression may be caused by an adrenaline secreting tumor. Dr. House is able to locate the tumor and diagnoses the patient with pheochromocytoma. Dr. Foreman, one of the doctors, attempts to appeal the inmate's death penalty on the basis that he was unable to control his actions due to his tumor. This kind of legal defense is rarely successful, however.
↑ 2.02.12.2"Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979". Mayo Clinic Proceedings58 (12): 802–4. December 1983. PMID6645626.
↑"What is the difference between pheochromocytoma and paraganglioma? What are the familial syndromes that have pheochromocytoma as a component? What are the pathologic features of pheochromocytoma indicating malignancy?". Questions in Daily Urologic Practice. Tokyo: Springer Japan. 2008. pp. 280–284. doi:10.1007/978-4-431-72819-1_49. ISBN978-4-431-72819-1.
↑"Pheochromocytomas and Paragangliomas". Endocrinology and Metabolism Clinics of North America48 (4): 727–750. December 2019. doi:10.1016/j.ecl.2019.08.006. PMID31655773.
↑"Life-threatening events in patients with pheochromocytoma". European Journal of Endocrinology173 (6): 757–64. December 2015. doi:10.1530/EJE-15-0483. PMID26346138.
↑"Pheochromocytoma and pregnancy: a deceptive connection". European Journal of Endocrinology166 (2): 143–50. February 2012. doi:10.1530/EJE-11-0528. PMID21890650.
↑ 20.020.1"Pheochromocytoma as a reversible cause of cardiomyopathy: Analysis and review of the literature". International Journal of Cardiology249: 319–323. December 2017. doi:10.1016/j.ijcard.2017.07.014. PMID29121733.
↑"Pheochromocytoma presenting as stroke in two Taiwanese children". Journal of Pediatric Endocrinology & Metabolism15 (9): 1563–7. November 2002. doi:10.1515/jpem.2002.15.9.1563. PMID12503867.
↑"Pheochromocytoma crisis resulting in acute heart failure and cardioembolic stroke in a 37-year-old man". Surgery155 (4): 726–7. April 2014. doi:10.1016/j.surg.2012.11.013. PMID23305592.
↑"Malignant paraganglioma presenting with hemorrhagic stroke in a child". Pediatrics132 (6): e1709-14. December 2013. doi:10.1542/peds.2013-0492. PMID24276837.
↑"Neurological manifestations of phaeochromocytomas and secretory paragangliomas: a reappraisal". Journal of Neurology, Neurosurgery, and Psychiatry84 (4): 452–7. April 2013. doi:10.1136/jnnp-2012-303028. PMID23204473.
↑"A case of pheochromocytoma complicated with acute renal failure and cardiomyopathy". Japanese Circulation Journal57 (1): 84–90. January 1993. doi:10.1253/jcj.57.84. PMID8437346.
↑"Pheochromocytoma presenting with rhabdomyolysis and acute renal failure: a case report". Renal Failure36 (1): 104–7. February 2014. doi:10.3109/0886022X.2013.832856. PMID24059440.
↑"Acute renal failure and transient, massive proteinuria in a case of pheochromocytoma". Clinical Nephrology24 (1): 47–9. July 1985. PMID4017298.
↑ 39.039.1"Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment". Hematology/Oncology Clinics of North America30 (1): 135–50. February 2016. doi:10.1016/j.hoc.2015.09.006. PMID26614373.
↑ 41.041.141.241.3"Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline". The Journal of Clinical Endocrinology and Metabolism99 (6): 1915–42. June 2014. doi:10.1210/jc.2014-1498. PMID24893135.
↑[non-primary source needed]"Presymptomatic genetic testing in minors at risk of paraganglioma and pheochromocytoma: our experience of oncogenetic multidisciplinary consultation". Hormone and Metabolic Research44 (5): 354–8. May 2012. doi:10.1055/s-0032-1311568. PMID22517555.
↑"65 YEARS OF THE DOUBLE HELIX: Genetics informs precision practice in the diagnosis and management of pheochromocytoma". Endocrine-Related Cancer25 (8): T201–T219. August 2018. doi:10.1530/ERC-18-0085. PMID29794110.
↑"Paraganglioma and phaeochromocytoma: from genetics to personalized medicine". Nature Reviews. Endocrinology11 (2): 101–11. February 2015. doi:10.1038/nrendo.2014.188. PMID25385035.
↑"Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity". Nature Reviews. Cancer14 (2): 108–19. February 2014. doi:10.1038/nrc3648. PMID24442145.
↑ 61.061.161.261.361.461.5"Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results". The Journal of Clinical Endocrinology and Metabolism88 (6): 2656–66. June 2003. doi:10.1210/jc.2002-030005. PMID12788870.
↑"Elevated urinary free and deconjugated catecholamines after consumption of a catecholamine-rich diet". The Journal of Clinical Endocrinology and Metabolism95 (6): 2851–5. June 2010. doi:10.1210/jc.2009-2589. PMID20382681.
↑"Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors". The Journal of Clinical Endocrinology and Metabolism94 (8): 2841–9. August 2009. doi:10.1210/jc.2009-0303. PMID19567530.
↑"[Diagnosis of pheochromocytoma and paraganglioma: the clonidine suppression test in patients with borderline elevations of plasma free normetanephrine]". Deutsche Medizinische Wochenschrift138 (3): 76–81. January 2013. doi:10.1055/s-0032-1327395. PMID23299341.
↑ 79.079.179.2"Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines". Clinical Chemistry51 (4): 735–44. April 2005. doi:10.1373/clinchem.2004.045484. PMID15718487.
↑"Reasons for inadequate or incomplete imaging techniques". Medical Ultrasonography20 (4): 498–507. December 2018. doi:10.11152/mu-1736. PMID30534659.
↑ 85.085.1"15 YEARS OF PARAGANGLIOMA: Imaging and imaging-based treatment of pheochromocytoma and paraganglioma". Endocrine-Related Cancer22 (4): T135-45. August 2015. doi:10.1530/ERC-15-0175. PMID26045470.
↑"Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging". AJR. American Journal of Roentgenology200 (2): 370–8. February 2013. doi:10.2214/AJR.12.9126. PMID23345359.
↑"The evolution in the use of MIBG scintigraphy in pheochromocytomas and paragangliomas". Hormones12 (1): 58–68. January 2013. doi:10.1007/bf03401287. PMID23624132.
↑"(131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis". Clinical Endocrinology80 (4): 487–501. April 2014. doi:10.1111/cen.12341. PMID24118038.
↑"Role of (18) F-FDOPA PET/CT imaging in endocrinology". Clinical Endocrinology81 (6): 789–98. December 2014. doi:10.1111/cen.12566. PMID25056984.
↑ 96.096.1"Peptide Receptor Radionuclide Therapy as a Novel Treatment for Metastatic and Invasive Phaeochromocytoma and Paraganglioma". Neuroendocrinology109 (4): 287–298. 2019. doi:10.1159/000499497. PMID30856620.
↑"Surgery for pheochromocytoma: A 20-year experience of a single institution". Hormones16 (4): 388–395. October 2017. doi:10.14310/horm.2002.1759. PMID29518759.
↑"Patterns of Use and Short-Term Outcomes of Minimally Invasive Surgery for Malignant Pheochromocytoma: A Population-Level Study". World Journal of Surgery39 (8): 1966–73. August 2015. doi:10.1007/s00268-015-3040-6. PMID25821949.
↑"Robotic posterior retroperitoneal adrenalectomy: operative technique". Archives of Surgery145 (8): 781–4. August 2010. doi:10.1001/archsurg.2010.148. PMID20713932.
↑"Both preoperative alpha and calcium channel blockade impact intraoperative hemodynamic stability similarly in the management of pheochromocytoma". Surgery156 (6): 1410–7; discussion1417-8. December 2014. doi:10.1016/j.surg.2014.08.022. PMID25456922.
↑"Beta-adrenergic blocking agents: their current status". AACN Clinical Issues in Critical Care Nursing3 (2): 447–60. May 1992. doi:10.4037/15597768-1992-2016. PMID1349490.
↑"Differential distribution of beta-adrenergic receptor subtypes in blood vessels of knockout mice lacking beta(1)- or beta(2)-adrenergic receptors". Molecular Pharmacology60 (5): 955–62. November 2001. doi:10.1124/mol.60.5.955. PMID11641423.
↑ 116.0116.1"Perioperative α-receptor blockade in phaeochromocytoma surgery: an observational case series". British Journal of Anaesthesia118 (2): 182–189. February 2017. doi:10.1093/bja/aew392. PMID28100521.
↑"Dogma is Made to be Broken. Why Are We Postponing Curative Surgery to Administer Ineffective Alpha Adrenoreceptor Blockade in Most Patients Undergoing Pheochromocytoma Removal?". Endocrine Practice25 (2): 199. February 2019. doi:10.4158/1934-2403-25.2.199. PMID30817194.
↑"Preoperative risk factors for haemodynamic instability during pheochromocytoma surgery in Chinese patients". Clinical Endocrinology88 (3): 498–505. March 2018. doi:10.1111/cen.13544. PMID29292527.
↑"Increased arterial pressure is not predictive of haemodynamic instability in patients undergoing adrenalectomy for phaeochromocytoma". Acta Anaesthesiologica Scandinavica53 (4): 522–7. April 2009. doi:10.1111/j.1399-6576.2008.01894.x. PMID19239408.
↑"Selective use of steroid replacement after adrenalectomy: lessons from 331 consecutive cases". Archives of Surgery141 (8): 771–4; discussion 774–6. August 2006. doi:10.1001/archsurg.141.8.771. PMID16924084.
↑"Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients With Metastatic Pheochromocytoma or Sympathetic Paraganglioma". Annals of Surgery268 (1): 172–178. July 2018. doi:10.1097/sla.0000000000002195. PMID28257320.
↑"Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma". Current Oncology Reports15 (4): 356–71. August 2013. doi:10.1007/s11912-013-0320-x. PMID23674235.
↑"Diagnosis and management of pheochromocytoma: a practical guide to clinicians". Current Hypertension Reports16 (7): 442. July 2014. doi:10.1007/s11906-014-0442-z. PMID24792093.
↑"I-123 MIBG imaging and intraoperative localization of metastatic pheochromocytoma: a case report". Clinical Nuclear Medicine27 (3): 183–5. March 2002. doi:10.1097/00003072-200203000-00007. PMID11852305.
↑"Management and outcome of metastatic pheochromocytomas/paragangliomas: an overview". Journal of Endocrinological Investigation44 (1): 15–25. June 2020. doi:10.1007/s40618-020-01344-z. PMID32602077.
↑ 145.0145.1"Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis". Clinical Endocrinology81 (5): 642–51. November 2014. doi:10.1111/cen.12542. PMID25041164.
↑"Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine". Annals of Internal Medicine109 (4): 267–73. August 1988. doi:10.7326/0003-4819-109-4-267. PMID3395037.
↑"Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy". The Journal of Clinical Endocrinology and Metabolism94 (8): 2850–6. August 2009. doi:10.1210/jc.2008-2697. PMID19470630.
↑"SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma". International Journal of Cancer135 (11): 2711–20. December 2014. doi:10.1002/ijc.28913. PMID24752622.
↑"Efficacy of Peptide Receptor Radionuclide Therapy for Functional Metastatic Paraganglioma and Pheochromocytoma". The Journal of Clinical Endocrinology and Metabolism102 (9): 3278–3287. September 2017. doi:10.1210/jc.2017-00816. PMID28605448.
↑"Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: A systematic review and meta-analysis". Clinical Endocrinology91 (6): 718–727. December 2019. doi:10.1111/cen.14106. PMID31569282.
↑"Changes in clinical features and long-term prognosis in patients with pheochromocytoma". American Journal of Hypertension13 (1 Pt 1): 35–43. January 2000. doi:10.1016/S0895-7061(99)00139-9. PMID10678269.
↑ 165.0165.1"Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study". The Journal of Clinical Endocrinology and Metabolism104 (6): 2367–2374. June 2019. doi:10.1210/jc.2018-01968. PMID30715419.
↑"Metastatic pheochromocytoma and paraganglioma: recent advances in prognosis and management". Current Opinion in Endocrinology, Diabetes and Obesity26 (3): 146–154. June 2019. doi:10.1097/med.0000000000000476. PMID30893083.
↑"Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005". Nature Clinical Practice. Endocrinology & Metabolism3 (2): 92–102. February 2007. doi:10.1038/ncpendmet0396. PMID17237836.
↑"Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series". Mayo Clinic Proceedings56 (6): 354–60. June 1981. PMID6453259.
↑"The epidemiology of pheochromocytoma: increasing incidence and changing clinical presentation. A population-based retrospective study 1977–2015". Endocrine Abstracts. 2017-05-03. doi:10.1530/endoabs.49.oc1.4. ISSN1479-6848.
↑"Pheochromocytoma and paraganglioma". Neuroendocrinology - Pathological Situations and Diseases. Progress in Brain Research. 182. Elsevier. 2010. pp. 343–73. doi:10.1016/s0079-6123(10)82015-1. ISBN978-0-444-53616-7.
↑"Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment". Surgical Oncology Clinics of North America25 (1): 119–38. January 2016. doi:10.1016/j.soc.2015.08.006. PMID26610778.












.jpg)





{kind=link}