Biology:CACNA1C
 Generic protein structure example |
Voltage-dependent L-type calcium channel subunit alpha-1C (also known as Cav1.2) is a protein that in humans is encoded by the CACNA1C gene.[1] Cav1.2 is a subunit of L-type voltage-dependent calcium channel.[2]
Structure and function
This gene encodes an alpha-1 subunit of a voltage-dependent calcium channel. Calcium channels mediate the influx of calcium ions (Ca2+) into the cell upon membrane polarization (see membrane potential and calcium in biology).[3]
The alpha-1 subunit consists of 24 transmembrane segments and forms the pore through which ions pass into the cell. The calcium channel consists of a complex of alpha-1, alpha-2/delta and beta subunits in a 1:1:1 ratio. The S3-S4 linkers of Cav1.2 determine the gating phenotype and modulated gating kinetics of the channel.[4] Cav1.2 is widely expressed in the smooth muscle, pancreatic cells, fibroblasts, and neurons.[5][6] However, it is particularly important and well known for its expression in the heart where it mediates L-type currents, which causes calcium-induced calcium release from the ER Stores via ryanodine receptors. It depolarizes at -30mV and helps define the shape of the action potential in cardiac and smooth muscle.[4] The protein encoded by this gene binds to and is inhibited by dihydropyridine.[7] In the arteries of the brain, high levels of calcium in mitochondria elevates activity of nuclear factor kappa B NF-κB and transcription of CACNA1c and functional Cav1.2 expression increases.[8] Cav1.2 also regulates levels of osteoprotegerin.[9]
CaV1.2 is inhibited by the action of STIM1.[10]
Regulation
The activity of CaV1.2 channels is tightly regulated by the Ca2+ signals they produce. An increase in intracellular Ca2+ concentration implicated in Cav1.2 facilitation, a form of positive feedback called Ca2+-dependent facilitation, that amplifies Ca2+ influx. In addition, increasing influx intracellular Ca2+ concentration has implicated to exert the opposite effect Ca2+ dependent inactivation.[11] These activation and inactivation mechanisms both involve Ca2+ binding to calmodulin (CaM) in the IQ domain in the C-terminal tail of these channels.[12] Cav1.2 channels are arranged in cluster of eight, on average, in the cell membrane. When calcium ions bind to calmodulin, which in turn binds to a Cav1.2 channel, it allows the Cav1.2 channels within a cluster to interact with each other.[13] This results in channels working cooperatively when they open at the same time to allow more calcium ions to enter and then close together to allow the cell to relax.[13]

Clinical significance
Relevance of CACNA1C in clinical disorders
Timothy Syndrome
Timothy Syndrome is a rare autosomal dominant disorder caused by rare heterozygous missense (non-synonymous) variants (mutations) in CACNA1C.[14] These variants are typically called 'gain of function' variants as their functional impact alters the excitation of the Cav1.2 channel.[15] The most frequent causative pathogenic variants for Timothy syndrome are p.G406R and p.G402S. There are two subtypes of Timothy syndrome: Type 1 and Type 2.[16] Timothy Syndrome Type 1 is caused by p.G406R in exon 8, with individuals presenting with prolonged QT, cardiac arrhythmia, neurodevelopmental delays, syndactyly, hypoglycaemia and hypotonia.[17] Individuals with Type 2 predominantly harbour p.G406R too, but, due to alternative splicing, this variant occurs in exon 8A. Timothy syndrome Type 2 has a similar phenotype to type 1 but also exhibits hip dysplasia.[18] Further variants have been linked to both syndromes.
LongQT Type 8
Alongside Timothy Syndrome, high-penetrance missense CACNA1C variants have also been noted in patients with LongQT Type 8, predominantly with no further extra-cardiac symptoms presenting.[19] LongQT Type 8 is a condition which is categorised by a prolonged QT interval, syncope and ventricular arrhythmias.[20] Although extra-cardiac features are not common, this could be due to underreporting.
Insufficient evidence: Brugada Syndrome
Although CACNA1C variants have been identified in Brugada Syndrome patients, the evidence for variants (such as p.A39V and p.G490R) causing genetic aetiology is disputed.[21][22][23] The link between Brugada Syndrome and CACNA1C variants is limited and predominantly consists of single-family cases with limited disease segregation.[24][25] There is currently insufficient evidence for the impact of CACNA1C variants on Brugada Syndrome, as currently corroborated by the Genomics England Panel App.[26][27]
Moderate/low impact variants
Large-scale genetic analyses have shown the possibility that CACNA1C is associated with bipolar disorder[28] and subsequently also with schizophrenia.[29][30][31] Also, a CACNA1C risk allele has been associated to a disruption in brain connectivity in patients with bipolar disorder, while not or only to a minor degree, in their unaffected relatives or healthy controls.[32] In a first study in Indian population, the Schizophrenia associated Genome-wide association study (GWAS) single nucelotide polymorphism (SNP) was found not to be associated with the disease. Furthermore, the main effect of rs1006737 was found to be associated with spatial abilityefficiency scores. Subjects with genotypes carrying the risk allele of rs1006737 (G/A and A/A) were found to have higher spatial ability efficiency scores as compared to those with the G/G genotype. While in healthy controls those with G/A and A/A genotypes were found to have higher spatial memory processing speed scores than those with G/G genotypes, the former had lower scores than the latter in schizophrenia subjects. In the same study the genotypes with the risk allele of rs1006737 namely A/A was associated with a significantly lower Align rank transformed Abnormal and involuntary movement scale (AIMS) scores of Tardive dyskinesia(TD).[33]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective Wikipedia articles. [§ 1]
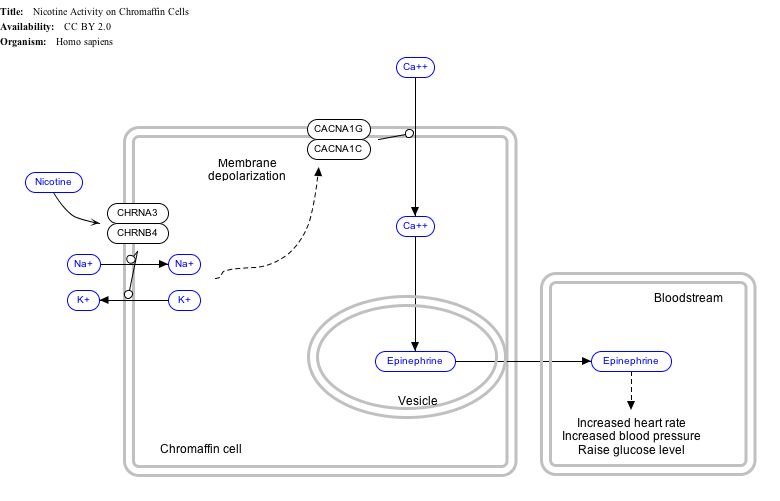
- ↑ The interactive pathway map can be edited at WikiPathways: "NicotineActivityonChromaffinCells_WP1603". http://www.wikipathways.org/index.php/Pathway:WP1603.
See also
References
- ↑ "Normalization of current kinetics by interaction between the alpha 1 and beta subunits of the skeletal muscle dihydropyridine-sensitive Ca2+ channel". Nature 352 (6335): 527–30. Aug 1991. doi:10.1038/352527a0. PMID 1650913. Bibcode: 1991Natur.352..527L.
- ↑ "International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels". Pharmacological Reviews 57 (4): 411–25. Dec 2005. doi:10.1124/pr.57.4.5. PMID 16382099.
- ↑ "L-type calcium channel targeting and local signalling in cardiac myocytes". Cardiovascular Research 98 (2): 177–86. May 2013. doi:10.1093/cvr/cvt021. PMID 23417040.
- ↑ 4.0 4.1 "L-type calcium channels: the low down". Journal of Neurophysiology 92 (5): 2633–41. Nov 2004. doi:10.1152/jn.00486.2004. PMID 15486420.
- ↑ "Ca2+-dependent modulation of voltage-gated Ca2+ channels". Biochimica et Biophysica Acta (BBA) - General Subjects 1820 (8): 1243–52. Aug 2012. doi:10.1016/j.bbagen.2011.12.012. PMID 22223119.
- ↑ "The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function". Cell and Tissue Research 357 (2): 463–76. Aug 2014. doi:10.1007/s00441-014-1936-3. PMID 24996399.
- ↑ "Entrez Gene: voltage-dependent, L type, alpha 1C subunit". https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=775.
- ↑ "Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries". Circulation Research 107 (5): 631–41. Sep 2010. doi:10.1161/CIRCRESAHA.110.224345. PMID 20616314.
- ↑ "Osteoprotegerin expression and secretion are regulated by calcium influx through the L-type voltage-sensitive calcium channel". Endocrinology 145 (1): 426–36. Jan 2004. doi:10.1210/en.2003-0319. PMID 14525906.
- ↑ "Cell biology. How to STIMulate calcium channels". Science 330 (6000): 43–4. Oct 2010. doi:10.1126/science.1196348. PMID 20929798.
- ↑ "Modulation of the voltage sensor of L-type Ca2+ channels by intracellular Ca2+". The Journal of General Physiology 123 (5): 555–71. May 2004. doi:10.1085/jgp.200308876. PMID 15111645.
- ↑ "Multiple C-terminal tail Ca(2+)/CaMs regulate Ca(V)1.2 function but do not mediate channel dimerization". The EMBO Journal 29 (23): 3924–38. Dec 2010. doi:10.1038/emboj.2010.260. PMID 20953164.
- ↑ 13.0 13.1 "Graded Ca2+/calmodulin-dependent coupling of voltage-gated CaV1.2 channels". eLife 4. 2015. doi:10.7554/eLife.05608. PMID 25714924.
- ↑ "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell 119 (1). October 2004. doi:10.1186/s13023-024-03445-x. PMID 15454078.
- ↑ "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell 119 (1): 19–31. October 2004. doi:10.1016/j.cell.2004.09.011. PMID 15454078.
- ↑ "Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development". American Journal of Medical Genetics. Part A 167A (3): 629–634. March 2015. doi:10.1002/ajmg.a.36924. PMID 25691416.
- ↑ "Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations". American Journal of Medical Genetics. Part A 173 (3): 784–789. March 2017. doi:10.1002/ajmg.a.38084. PMID 28211989.
- ↑ "A Natural History Study of Timothy Syndrome". Orphanet Journal of Rare Diseases 19 (1): 433. November 2024. doi:10.1186/s13023-024-03445-x. PMID 39580446.
- ↑ "Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome". Circulation. Cardiovascular Genetics 6 (3): 279–289. June 2013. doi:10.1161/CIRCGENETICS.113.000138. PMID 23677916.
- ↑ "Long QT syndrome type 8: novel CACNA1C mutations causing QT prolongation and variant phenotypes". Europace 16 (12): 1828–1837. December 2014. doi:10.1093/europace/euu063. PMID 24728418.
- ↑ "Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death". Circulation 115 (4): 442–449. January 2007. doi:10.1161/CIRCULATIONAHA.106.668392. PMID 17224476.
- ↑ "Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death". European Heart Journal 43 (15): 1500–1510. April 2022. doi:10.1093/eurheartj/ehab687. PMID 34557911.
- ↑ "Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome". Circulation 138 (12): 1195–1205. September 2018. doi:10.1161/CIRCULATIONAHA.118.035070. PMID 29959160.
- ↑ "Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations". Basic Research in Cardiology 109 (6). November 2014. doi:10.1007/s00395-014-0446-5. PMID 25341504.
- ↑ "Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death". Heart Rhythm 7 (12): 1872–1882. December 2010. doi:10.1016/j.hrthm.2010.08.026. PMID 20817017.
- ↑ "Role of CACNA1C in Brugada syndrome: Prevalence and phenotype of probands referred for genetic testing" (in English). Heart Rhythm 19 (5): 798–806. May 2022. doi:10.1016/j.hrthm.2021.12.032. PMID 34999275.
- ↑ "Role of CACNA1C variants in Brugada syndrome: clinical aspects and genetic testing strategies" (in en). European Heart Journal 41 (Supplement_2). 2020-11-01. doi:10.1093/ehjci/ehaa946.3584. ISSN 0195-668X. https://academic.oup.com/eurheartj/article/doi/10.1093/ehjci/ehaa946.3584/6003560.
- ↑ "Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder". Nature Genetics 40 (9): 1056–8. Sep 2008. doi:10.1038/ng.209. PMID 18711365.
- ↑ "The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia". Molecular Psychiatry 15 (10): 1016–22. Oct 2010. doi:10.1038/mp.2009.49. PMID 19621016.
- ↑ "Case-case genome-wide association analysis shows markers differentially associated with schizophrenia and bipolar disorder and implicates calcium channel genes". Psychiatric Genetics 21 (1): 1–4. Feb 2011. doi:10.1097/YPG.0b013e3283413382. PMID 21057379.
- ↑ Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014-07-24). "Biological insights from 108 schizophrenia-associated genetic loci". Nature 511 (7510): 421–427. doi:10.1038/nature13595. ISSN 1476-4687. PMID 25056061. Bibcode: 2014Natur.511..421S.
- ↑ "The impact of CACNA1C allelic variation on effective connectivity during emotional processing in bipolar disorder". Molecular Psychiatry 18 (5): 526–7. May 2013. doi:10.1038/mp.2012.61. PMID 22614292.
- ↑ "Effect of rs1108580 of DBH and rs1006737 of CACNA1C on Cognition and Tardive Dyskinesia in a North Indian Schizophrenia Cohort". Molecular Neurobiology 60 (12): 6826–6839. 2023. doi:10.1007/s12035-023-03496-4. PMID 37493923.
Further reading
- "Effects of the CACNA1C risk allele for bipolar disorder on cerebral gray matter volume in healthy individuals". The American Journal of Psychiatry 166 (12): 1413–4. Dec 2009. doi:10.1176/appi.ajp.2009.09050680. PMID 19952088.
- "Molecular diversity of L-type Ca2+ channel transcripts in human fibroblasts". Proceedings of the National Academy of Sciences of the United States of America 89 (10): 4628–32. May 1992. doi:10.1073/pnas.89.10.4628. PMID 1316612. Bibcode: 1992PNAS...89.4628S.
- "Linkage mapping of the human gene for the alpha 1 subunit of the cardiac DHP-sensitive Ca2+ channel (CACNL1A1) to chromosome 12p13.2-pter using a dinucleotide repeat". Genomics 14 (1): 206–7. Sep 1992. doi:10.1016/S0888-7543(05)80312-X. PMID 1330882.
- "Mapping of a human brain voltage-gated calcium channel to human chromosome 12p13-pter". Genomics 14 (4): 1092–4. Dec 1992. doi:10.1016/S0888-7543(05)80135-1. PMID 1335957.
- "Assignment of the human gene for the alpha 1 subunit of the cardiac DHP-sensitive Ca2+ channel (CCHL1A1) to chromosome 12p12-pter". Genomics 10 (3): 835–9. Jul 1991. doi:10.1016/0888-7543(91)90471-P. PMID 1653763.
- "Molecular diversity of L-type calcium channels. Evidence for alternative splicing of the transcripts of three non-allelic genes". The Journal of Biological Chemistry 265 (33): 20430–6. Nov 1990. doi:10.1016/S0021-9258(17)30522-7. PMID 2173707.
- "Different voltage-dependent inhibition by dihydropyridines of human Ca2+ channel splice variants". The Journal of Biological Chemistry 270 (18): 10540–3. May 1995. doi:10.1074/jbc.270.18.10540. PMID 7737988.
- "Genomic structure of human L-type Ca2+ channel". Genomics 22 (1): 77–87. Jul 1994. doi:10.1006/geno.1994.1347. PMID 7959794.
- "Molecular localization of ion selectivity sites within the pore of a human L-type cardiac calcium channel". The Journal of Biological Chemistry 268 (18): 13026–9. Jun 1993. doi:10.1016/S0021-9258(19)38613-2. PMID 8099908.
- "Cloning, chromosomal localization, and functional expression of the alpha 1 subunit of the L-type voltage-dependent calcium channel from normal human heart". Proceedings of the National Academy of Sciences of the United States of America 90 (13): 6228–32. Jul 1993. doi:10.1073/pnas.90.13.6228. PMID 8392192. Bibcode: 1993PNAS...90.6228S.
- "A potential site of functional modulation by protein kinase A in the cardiac Ca2+ channel alpha 1C subunit". FEBS Letters 384 (2): 189–92. Apr 1996. doi:10.1016/0014-5793(96)00303-1. PMID 8612821. Bibcode: 1996FEBSL.384..189P.
- "A "double adaptor" method for improved shotgun library construction". Analytical Biochemistry 236 (1): 107–13. Apr 1996. doi:10.1006/abio.1996.0138. PMID 8619474.
- "Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40-42 in alpha1C subunit in the kinetics and Ca2+ dependence of inactivation". The Journal of Biological Chemistry 272 (6): 3560–6. Feb 1997. doi:10.1074/jbc.272.6.3560. PMID 9013606.
- "Properties of three COOH-terminal splice variants of a human cardiac L-type Ca2+-channel alpha1-subunit". The American Journal of Physiology 272 (3 Pt 2): H1372–81. Mar 1997. doi:10.1152/ajpheart.1997.272.3.H1372. PMID 9087614.
- "Large-scale concatenation cDNA sequencing". Genome Research 7 (4): 353–8. Apr 1997. doi:10.1101/gr.7.4.353. PMID 9110174.
- "cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits". Neuron 19 (1): 185–96. Jul 1997. doi:10.1016/S0896-6273(00)80358-X. PMID 9247274.
- "Ca2+ channel sensitivity towards the blocker isradipine is affected by alternative splicing of the human alpha1C subunit gene". FEBS Letters 427 (2): 220–4. May 1998. doi:10.1016/S0014-5793(98)00425-6. PMID 9607315. Bibcode: 1998FEBSL.427..220Z.
- "Sorcin associates with the pore-forming subunit of voltage-dependent L-type Ca2+ channels". The Journal of Biological Chemistry 273 (30): 18930–5. Jul 1998. doi:10.1074/jbc.273.30.18930. PMID 9668070.
- "Fiber-FISH analysis of the 3'-terminal region of the human L-type Ca2+ channel alpha 1C subunit gene". Hereditas 129 (2): 169–75. 1999. doi:10.1111/j.1601-5223.1998.00169.x. PMID 10022083.
External links
- GeneReviews/NIH/NCBI/UW entry on Brugada syndrome
- CACNA1C+protein,+human at the US National Library of Medicine Medical Subject Headings (MeSH)
- GeneReviews/NIH/NCBI/UW entry on Timothy Syndrome
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|

 |  |


{kind=link}